Now that I have my new MacPro I thought it might be interesting to try out a couple of the software packages that I’ve previously reviewed. ForgeV10 allows the scientist to use Cresset’s proprietary electrostatic and physicochemical fields to align, score and compare diverse molecules. It allows the user to build field based pharmacophores to understand structure activity and then use the template to undertake a virtual screen to identify novel scaffolds. I’ve previously reviewed ForgeV10 and as it was formally known FieldAlign so I’m going to focus on the support for multiple processors and a few of the new features.
Multi-core Support
It is clear that we are not going to see major improvements in chip speed so to improve performance many scientific packages have taken advantage of the parallel processing capabilities of modern multicore chips. Indeed it could be argued that any scientific software developer not looking to exploit the modern chip architectures is not doing there users any favours.
This is the configuration of my new desktop machine. Mac Pro Z0P8 Configuration:
- 3.5GHz 6-core with 12MB of L3 cache
- 64GB (4 x 16GB) of 1866MHz DDR3 ECC
- 512GB PCIe-based flash storage
- Dual AMD FirePro D700 GPUs with 6GB of GDDR5 VRAM each
In the Forge preferences you can set the number of processors to use so I set it to 6 and then did a test run using a PDE4 test set. Rolipram is a known PDE4 inhibitor and there is an X-ray structure of the protein in the public domain 1RO6 I downloaded this structure and cleaned it up using MOE then saved Rolipram and the protein as mol2 files, I also had a file containing around 200 potential ligands as 2D structures in sdf file format. The test involved both conformation searching, minimisation and alignment in the protein. If you start with a file containing pre-computed conformations then you can omit first step. However in my experience the Cresset tools do a very good job at generating reasonable conformations, much better than many alternative tools.
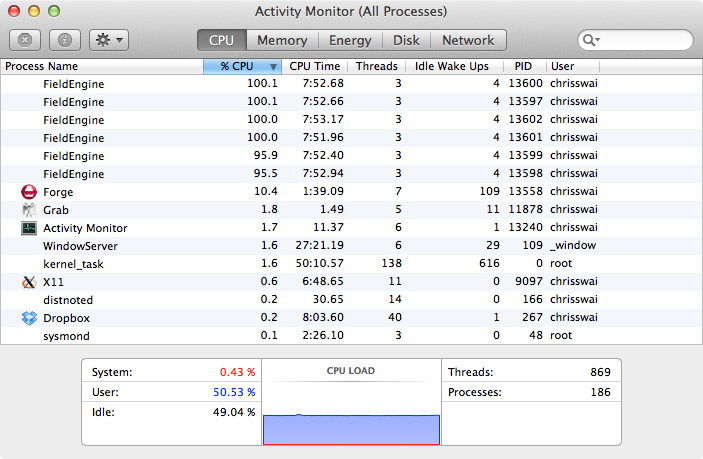
Looking at Activity Monitor (above) we can see the 6 FieldEngine processes running at approx 100%, but it was clear that the CPU load was only around 50%. Talking to the excellent Cresset support it was clear that there was the opportunity to take advantage of hyper-threading
For each processor core that is physically present, the operating system addresses two virtual or logical cores, and shares the workload between them when possible. The main function of hyper-threading is to decrease the number of dependent instructions on the pipeline. It takes advantage of superscalar architecture (multiple instructions operating on separate data in parallel). They appear to the OS as two processors, thus the OS can schedule two processes at once. http://en.wikipedia.org/wiki/Hyper-threading.
So an an experiment I ran the same dataset but changed the number of cores used over the range 3-8 (8 is the maximum number of threads supported by Forge) in the Forge preferences. As you can see in the histogram below increasing the number of cores available reduces the time taken to process the structure file.
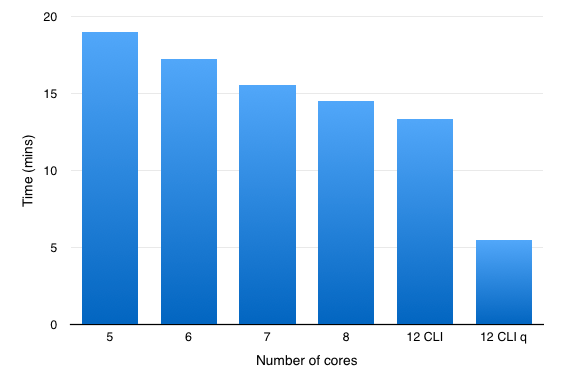
You can also access the falign module via the command line interface (CLI), and for this comparison I used
/Applications/Forge.app/Contents/MacOS/falign /Users/username/Desktop/ForForge/rolipram.mol2 /Users/username/Desktop/ForForge/for_PFDE4_search200.sdf -p /Users/username/Desktop/ForForge/protein.mol2 > /Users/username/Desktop/ForForge/results.sdf
Looking at Activity Monitor is was apparent that 12 FieldEngines were initiated.
I also tried using the -q option
/Applications/Forge.app/Contents/MacOS/falign /Users/username/Desktop/ForForge/rolipram.mol2 /Users/username/Desktop/ForForge/for_PFDE4_search200.sdf -q -p /Users/username/Desktop/ForForge/protein.mol2 > /Users/username/Desktop/ForForge/results.sdf
The Command Line Interface
I have to say the help is probably the clearest and most comprehensive I have seen for a command line application!
/Applications/Forge.app/Contents/MacOS/falign -H
Usage: falign [options] reference_file [db_file1] [db_file2] ...
falign [options] project_file.fqj [db_file1] [db_file2] ...
falign [options] project_file.fpj [db_file1] [db_file2] ...
This program reads in a reference molecule or set of molecules. It then reads another series of molecules. Each of these has conformers generated if required, and is then aligned to the reference molecules. The final set of aligned conformations is then written to standard output.
Alternatively, the reference molecule and molecules to be aligned can be read from a Forge project file specified as the first file argument. All other relevant data from that project file (e.g. field constraints and protein) will be used as well. If the -u option is given, then the full set of calculation parameters last used on that project will be loaded and used.
Options:
========
-a, --alignments A
Output the top A alignments for each molecule (default 1).
-c, --coulombics
Use full coulombics and van der Waals interactions during the conformer generation. The default is to neglect coulombics and attractive vdW forces, which usually gives a better conformer population.
-C, --charge
Assign protonation state for all molecules to be aligned for pH 7.
-e, --exhaustive
Generate more conformers and spend more CPU time on the alignments.
-E, --engine path
Set the path to the 'FieldEngine' binary for local processing.
-f, --fieldconstraint index,size
Specifies a field constraint on the field point with the specified index. Multiple constraints can be specified, or multiple -f options can be given.
-F, --filter filterRMS
Set the RMS filter for conformer generation: two conformers are treated as being different if they are at least this far apart. The default is 0.5A, (or 0.7A if
-q is specified).
-h, --help
Print short help text.
-H, --extendedhelp
Print this help text.
-i, --input xed|sdf|mol2|smi
Set the file format for reading the molecules to be aligned. The default is to autodetect when reading from files, or to assume SDF files when reading from standard input.
-I, --immobile
Do not generate conformations or allow molecules to move, just score molecules
in their input positions.
-k, --keep-all-confs
By default, only confomers that are used in alignments are kept. If this option is set, all conformers will be kept. This only has an effect if a project file is being written with the -P option. On 32-bit platforms, specifying this option will limit the number of molecules that can be processed.
-l, --localengines numEngines
Set the number of local engines to start. Use 0 for no local processing.
-L, --log
Request that the calculation log be written as one of the SDF tags for each alignment. Only has an effect if '-o sdf' (the default) is active.
-m, --maxconfs M
Create at most M conformers per molecule. The default is 100, 50 with the -q switch, and 200 with the -e switch.
-M, --mcs normal/skeleton/flexible
Generate conformers and alignments by performing a maximum-common-substructure match to the references, and generating conformers by moving only the atomsoutside the MCS. Specify the MCS matching rule to use: 'normal' means match on element+hybridisation, 'skeleton' means match on hybridisation only, and 'flexible' matches on hybridisation only and allows ring and chain atoms to match. You can specify 'n'/'s'/'f' as shortcuts for the arguments.
-n, --no-invert
Do not allow conformers of achiral molecules to be inverted if that gives a better alignment. Chiral molecules are never inverted. Specify this only when you are reading in conformer populations generated by an external program which does output conformational enantiomers.
-N, --networkengines hostname:port,hostname:port,...
Give the locations of additional engine processes. Alternatively, the locations can be provided in a file, using -N @filename. If using the @filename form, the engine locations can be separated by a comma or a newline.
-o, --output xed|sdf|mol2
Set the file format for the output. The default is to output SDF files.
-p, --protein protein_file
Read the first molecule in the specified file and use it as an excluded volum when aligning molecules.
-P, --project project_file
Write the results to the specified file as a Forge project which can be read by the GUI version. A '.fqj' extension will be added to the filename if not specified.
-q, --quick
Take shortcuts in the conformer generation and alignment: faster but possibly poorer alignments.
-r, --readmode molecules|conformers|fixed_confs|aligned_confs
Set how the database molecules should be read. In 'm'olecules mode, every entry is assumed to be a separate molecule that will require conformer generation. In 'c'onformers mode, each input file is assumed to hold multiple conformers of the same molecule i.e. one molecule per file, possibly as multiple confs. In 'f'ixed mode, each entry is assumed to be a separate molecule in a fixed conformation: it will require alignment but not conformer generation. In 'a'ligned mode, each entry is taken to be a separate molecule in a fixed orientation, requiring neither conformer generation nor alignment. The default is to autodetect between 'm' and 'c' depending on the file contents.
-R, --protein-hardness soft|medium|hard
Set the hardness of the protein excluded volume to use: a harder protein penalises steric clashes more. Only has an effect if -p is specified.
-s, --shapewt S
Set the weight of the shape component of the alignment function. The default, 0.5, means half shape and half fields.
-S, --mcs-static
Once an MCS alignment has been performed, the aligned molecule may not move. The default is to allow the aligned molecule to move slightly as a rigid body to maximise the alignment score. Only has an effect if -M is specified.
-u, --use-stored-settings
If a project file was specified as one of the inputs, load the calculation settings to use from that project file. The options affected are -c, -e, -f, -F, -k, -m, -M, -n, -R, -s, -S and -x.
-v, --verbose
Print progress information. Multiple -vs increase verbosity.
-V, --version
Print version and license information.
-w, --writeref before/after
Write the reference molecule out before or after the alignments.
-W, --weightAtoB wt
Set the reference-fieldpoints-into-db-field weight. The default, 0.5, means that the reference and the database molecule's field points have equal weight.
-x, --max-score
If there is more than one reference molecule, then each molecule's score is taken to be the best score to any individual reference. The default is to use the average score among all of the references.
From my results a back of the envelope calculation would suggest my machine would be capable of processing around 21,000 structures per day (100 conformations, 10 alignments) using the GUI to set up the calculation. Whilst the figures will vary depending on the protein and the small molecule structures this is a very impressive throughput. This would be ideal for looking at the hits from a high-throughput screen, or one could imagine using a docking program to rapidly evaluate a few million structures and then take the top 20-30 thousand structures and using Forge to provide an alternative evaluation. It could also be useful for evaluating all compounds from a ongoing MedChem project, or looking at the results from an off-target activity. Using the command line interface (CLI) it is possible to make use of more FieldEngines and to tune the calculation. For instance using -q option to reduce the number of conformations generated and faster alignments.
I should emphasise ForgeV10.2 is not a virtual screening tool, Cresset have another product Blaze that has been optimised for very high-throughput virtual screening. If you have ever looked at virtual screening results you will be aware of the strange conformations and unlikely interactions that can sometimes be found, ForgeV10.2 identifies high quality conformations and gives much more believable results, the improved quality does come at a computational cost but it certainly seems like the new MacPro is up to the task.
Other New Features
Version 10.2 brings the inclusion of Activity Miner that I’ve previously reviewed, Activity Miner from Cresset is a new tool designed to rapidly interrogate and decipher SAR in both Torch and Forge. Activity Miner is intended to help identify key elements of the SAR by starting from a set of aligned molecules and then automatically comparing them to each other. Each pair is given a ‘disparity’ value which reflects how much the activity changes relative to the structure.
There are also a number of bug fixes and minor tweaks including the addition of circular fingerprints (ECFP4) and circular pharmacophore fingerprints (FCFP6) as options for the 2D similarity method. Ligand Efficiency and Lipophillic Ligand Efficiency columns have been added to the Results table
Last updated 7 May 2014