ForgeV10 is the latest offering from Cresset, whilst a new product those familiar with FieldAlign and FieldTemplater will recognise much of the functionality. ForgeV10 allows the scientist to use Cresset’s proprietary electrostatic and physicochemical fields to align, score and compare diverse molecules. It allows the user to build field based pharmacophores to understand structure activity and then use the template to undertake a virtual screen to identify novel scaffolds.
Background
Molecules acting at the same active site usually do so because they have similar electrostatic and van der Waals surfaces in the bound conformation, and this can be irrespective of the underlying molecular framework. Molecular connectivity descriptors have proved to be useful for identifying similar ligands but are not ideal for identifying novel structures with similar shape and electrostatics with a different scaffold. A comparison of the complete surface properties for all conformations would be a hugely demanding computational task so instead Cresset use field points, representing the maxima and minima of the fields. These field points are then used for rapid comparison of structures. The field points are actually generated by calculating the interaction energy with a probe atom. Blue are negative field points, red positive and orange hydrophobic. The relative size of the points reflects the size of the interaction energy.
2D Representation
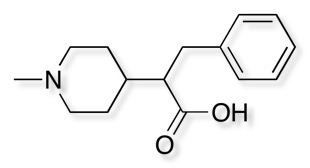
3D with Field Points added
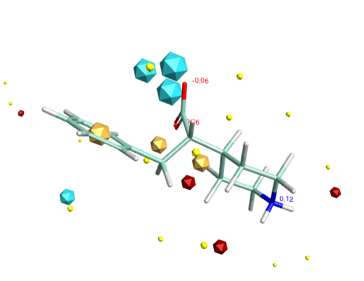
3D with electrostatic surfaces (Blue negative, Red positive)
Using ForgeV10
Forge can be used in two modes. If the 3D shape of the ligands bound to the active site of the target is known then it takes a single or small set of molecules in a predefined conformation to use as a “reference”. It then aligns a series of database molecules to the reference based on molecular fields, shape or common substructure to the reference. On the other hand if the shape of the molecules in the protein is not known then the separate FieldTemplater module will cross compare diverse actives to give a hypothesis for how the molecules might relate in 3D when bound to the active site of the target.
Finding PDE4 ligands
Rolipram is a known PDE4 inhibitor and there is an X-ray structure in the public domain 1RO6 I downloaded this structure and cleaned it up using MOE then saved Rolipram and the protein as mol2 files, I also had a file containing potential ligands as 2D structures in sdf file format.
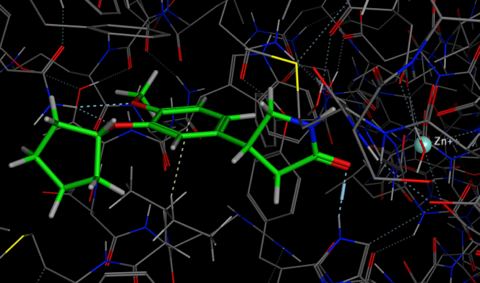
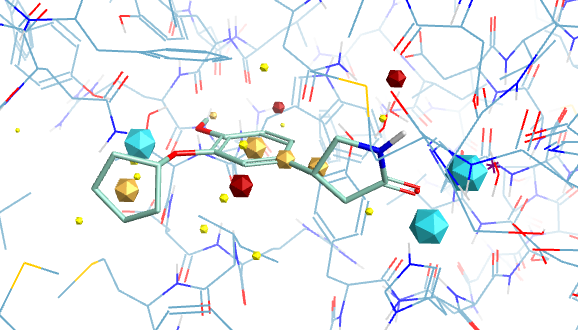
The easiest way to use Forge is to simply use the wizard to guide you through the process. The wizard first asks for the known template ligand(s) in this case I selected the Rolipram.mol2 and then the file containing the structures to be aligned and scored, thirdly we can select the protein structure which is simply used as an excluded volume in that any alignment that places one or more atoms inside this volume will be penalised. The penalty is proportional to the number of atoms entering the volume thus small infringements into the region do not significantly affect the alignment score. It is also possible to add field constraints if certain points are known to be critical. The next dialog gives you the option to influence the speed or accuracy of the subsequent calculation, in my experiments the conformational searching had the greatest influence on the time taken. If you are using small relatively rigid molecules it may be worth reducing the conformational search, however if the ligands contain medium sized rings it would be worth increasing the time.
The images below give an idea of the key field point interactions for Rolipram.
Field Points
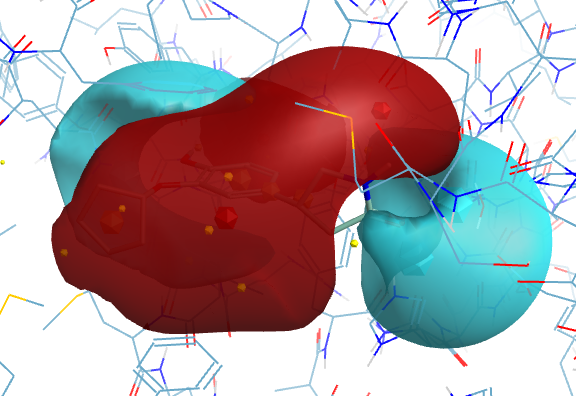
With electrostatic surfaces (Blue negative, Red positive)
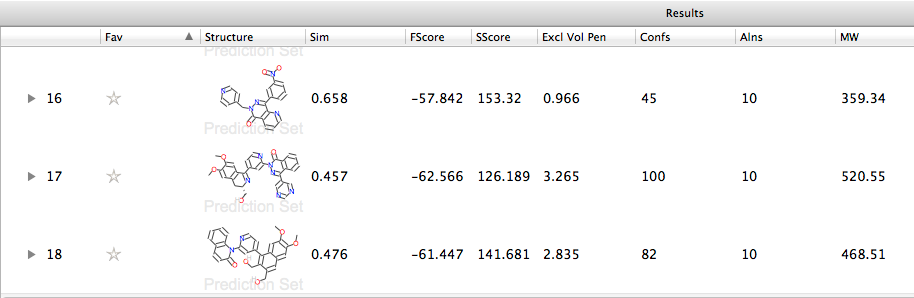
After running the calculation the results are displayed in a spreadsheet, giving the similarity score, but by right-clicking on the field titles you can alter the fields displayed, allowing you to compare the field and steric scores together with the excluded volume penalty due to interaction with the protein.
I exported the results as an sdf file and then imported the file into Vortex, I then calculated the similarity of each of the ligands to Rolipram using MACCS descriptors by running this Vortex script. The results were then plotted as shown below. On the x-axis we have the Forge similarity score on the vertical axis the MACCS Tanimoto similarity measurement, the points are colour coded according to the measured PDE4 activity (red most active). Taking 0.7 as the cut-off, it is clear very few of the active ligands are identified using MACCS descriptors (above red line), in contrast a considerable number of actives are identified using Forge (to the right of the blue line).
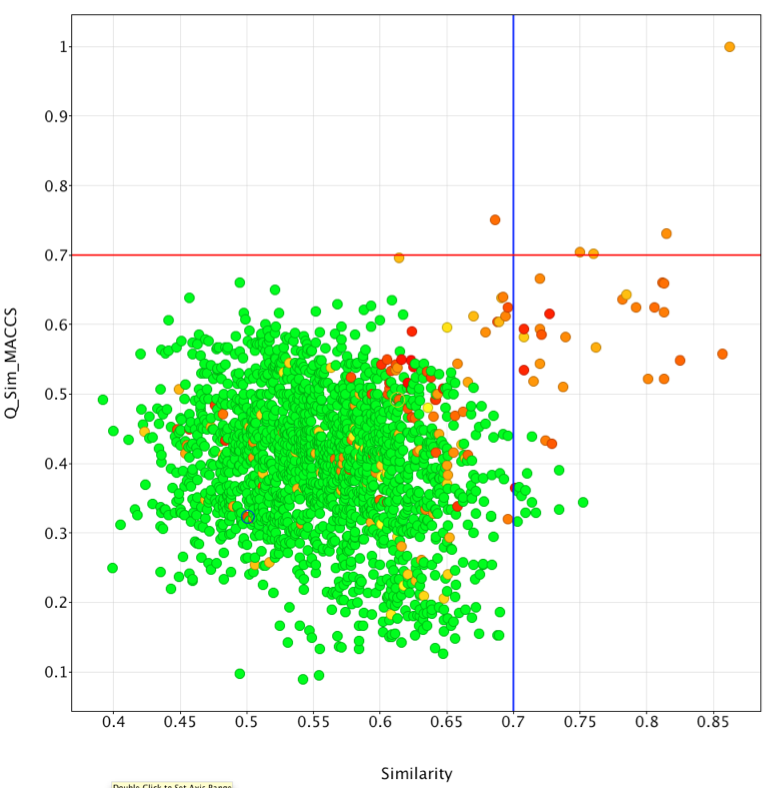
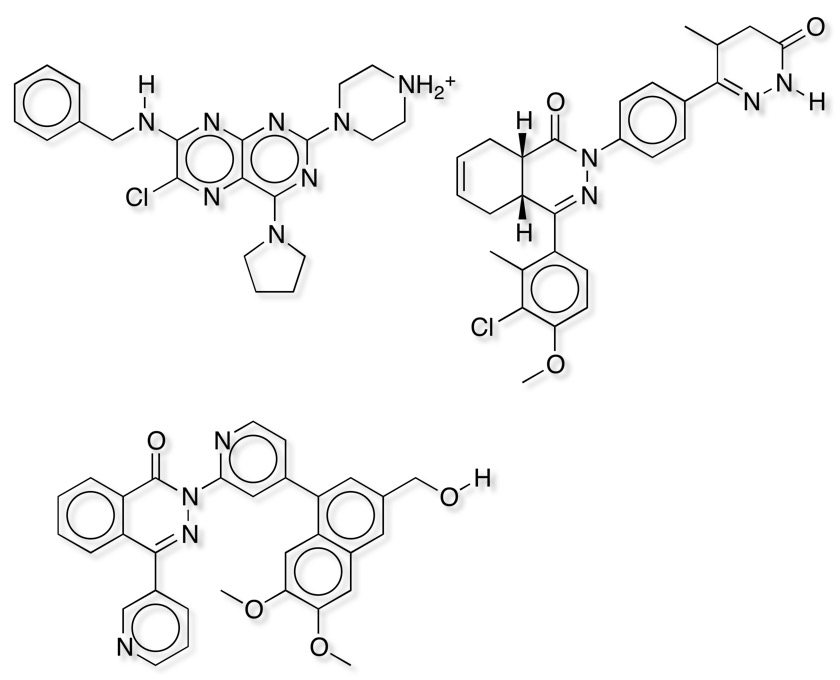
It is clear that both similarity measures fail to identify some potent ligands shown below, in general it seems that these compounds are much larger than the reference ligand Rolipram. It is important to note that Forge uses the protein only as an excluded volume, currently it will not identify any possible favourable new interactions between novel ligands and the protein that are not present in the reference molecule.
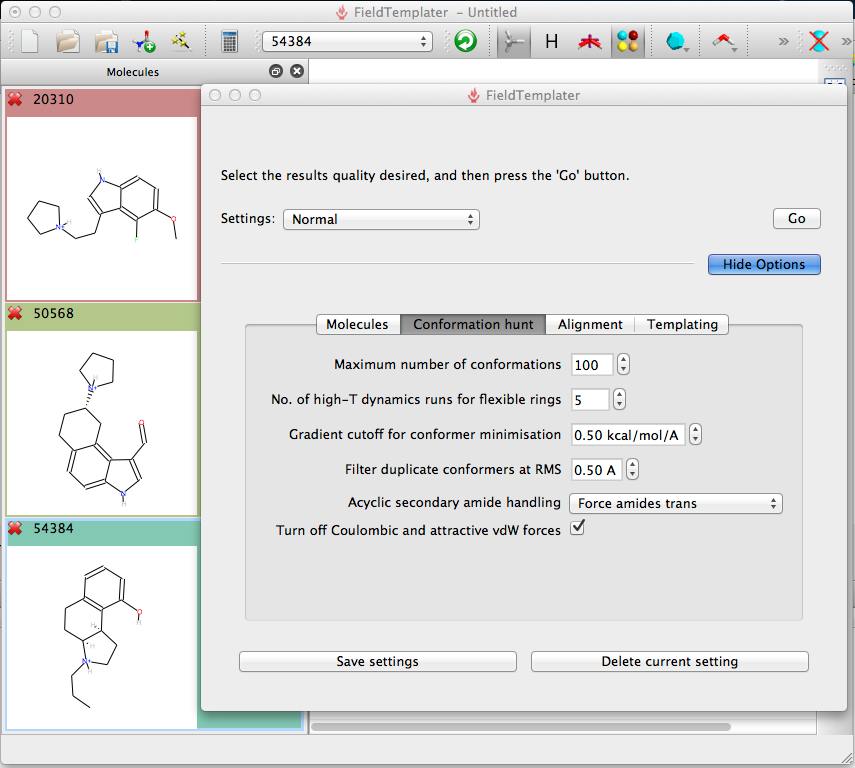
I did some experiments changing the various options and found that the conformation hunt perhaps has the greatest impact on the results, this was particularly true for compounds that contained medium sized rings, in these cases I found that increasing the number of high-T dynamics runs gave much better results. The other factor that can cause problems are occasions where there is the possibility of intramolecular hydrogen bonds or other intramolecular non-bonded interactions that might stabilise a particular interaction. There is an option to include Coulombic and attractive vdW forces but I did not explore it. There is also an option to specifically match a maximum common substructure, however in the examples I used substructures were overlaid and scored highly already.
In many cases three dimensional information about the protein target is not be available, but if a there is a group of known actives then FieldTemplater can be used to generate a hypothetical binding model and this used to screen. The FieldTemplater module searches for common field patterns across the explored conformational space of a set of ligands. A field pattern which can be generated by multiple independent structurally-distinct molecules is likely to be related to how those compounds bind to a common receptor.
The manual for FieldTemplater is in the application bundle (/Applications/Forge.app/Contents/Resources/FieldTemplater_10.0.0.pdf ) but not linked anywhere.
In my experiments the choice of molecules that are chosen to create the FieldTemplate is critical. I found that molecules that appear to bind to the same site based on a binding assay may actually use very different interactions with the target protein, In these cases I found FieldTemplater did not find a useful solution and in fact I got much better results by screening each of the compounds individually and then combining the results. I also found that very flexible ligands may cause difficulties so if possible choose the most conformationally rigid analogues. The size of the molecule also has an influence, the program assumes that all parts of the ligand are equally important, so if you have a phenyl ring that does not actually increase affinity then choose an analogue with no phenyl of the scoring will penalise molecules that do not occupy the volume occupied by the phenyl. FieldTemplater will accommodate upto 10 molecules but I got the best results with just 3 or 4 molecules. It is also important to include the active enantiomer.
After importing the molecules the user is presented with a control panel to simply accept the default setting, however displaying the options allows you to modify the settings. I suspect the experienced user will create their own preferred settings depending on the type of ligands, for flexible molecules you may want to increase the amount of conformational searching.
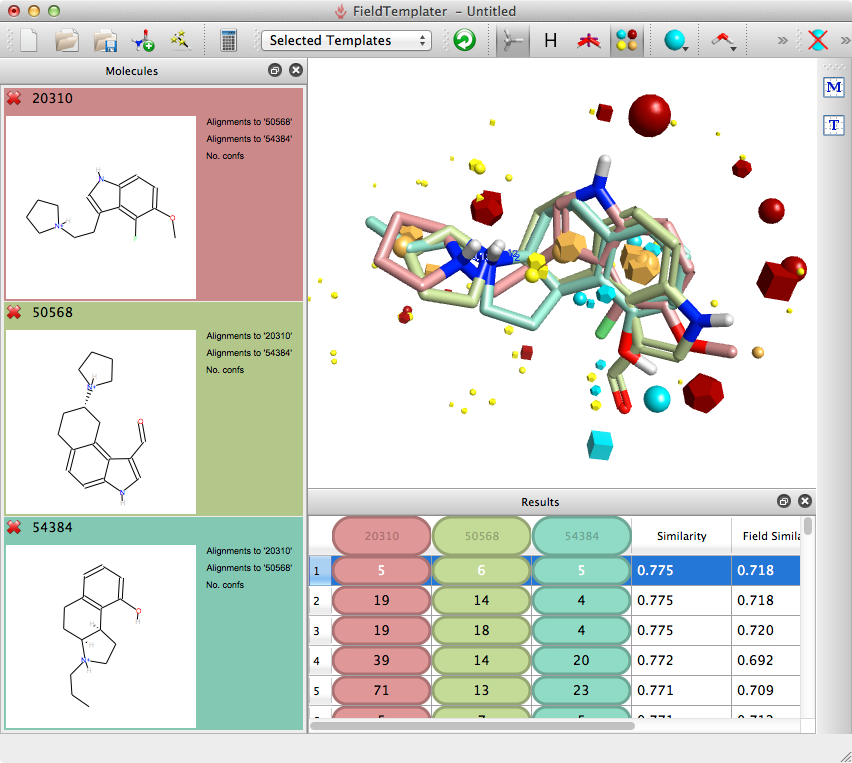
Generating the Field Template for the ligands above took less than 20 mins on my laptop, it is probably worth spending a little extra time here to refine the model since the searching of the database to predict and score is likely to be slower.
Templater will identify multiple overlays and it is worthwhile examining them before choosing the template to use for searching, especially if any SAR is not accounted for by the reference molecules.
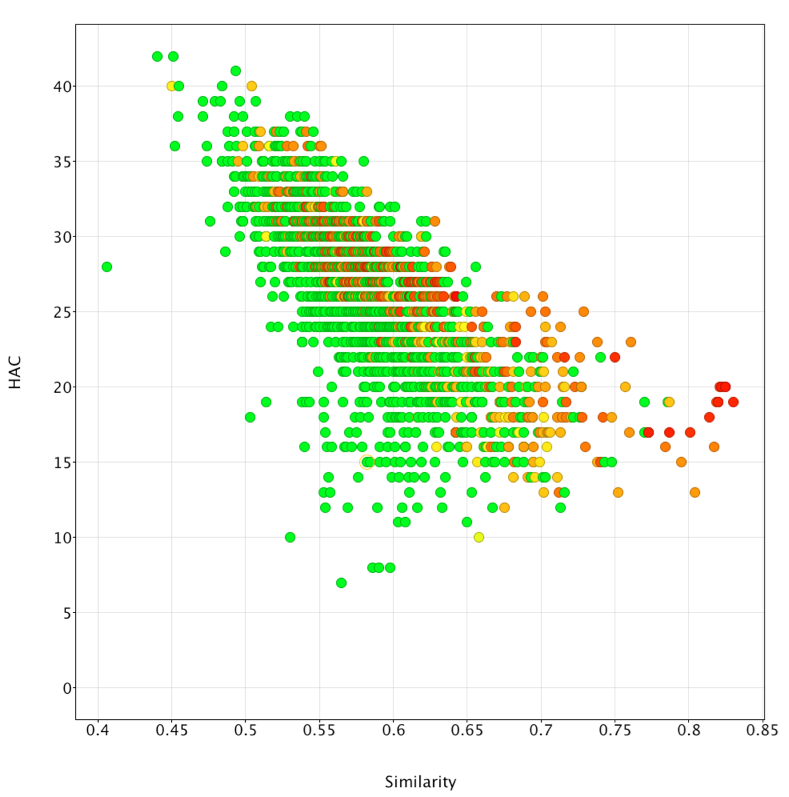
The desired template can then be copied into Forge and the similarity search undertaken. The results were exported as an sdf file and imported into Vortex for analysis, the plot below shows similarity versus heavy atom count (HAC), colour coded for activity (red active to green inactive). It is apparent that Forge does an excellent job of identifying active ligands that are similar in size to the ligands used to generate the template (around 20 heavy atoms) but is much less effective in identifying actives from much larger ligands.
External Models
Forge also has an option to score molecules based on external models via a plugin, in particular in can use models generated by Optibrium’s Automodeller and generate new columns based on the predicted activity in a variety of ADME models (these models were discussed in the StarDrop review).
Summary
ForgeV10 is an excellent tools for evaluating compounds and understanding SAR, to get the best from the software it probably needs to be in the hands of an experienced scientist, rather than the occasional bench medicinal chemist user. However it is possible to share templates and Field models for others to test their own ideas.
One thought on “A Review of ForgeV10”
Comments are closed.