In an earlier script I described the use of the ability to script multiple sub-structure searches using SMARTS. There are many occasions when this sort of feature is useful, if you want to flag molecules that contain reactive functional groups, toxicophores, or PAINS functional groups that have been shown to interfere with a variety of screens. Alternatively if you have a drug discovery project with multiple chemotypes you might want to tag particular groups of compounds as belonging to a named series to aid analysis.
Whilst the original script was certainly very useful it was rather slow on large workspaces, to calculate all the PAINS filters for the 1.4M structures in ChEMBL took around 1 hour. Over the last couple of months I’ve been working with the Dan Ormsby of Dotmatics to improve the speed of searching. The latest builds (greater than 37278) of Vortex contain a new multi-core cheminformatics engine that can be enabled in the preferences panel that greatly improves search speeds. I always think one of the best ways to evaluate new features is really stress test them!
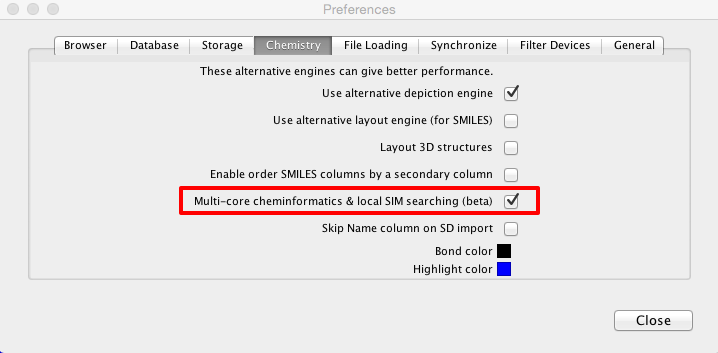
The results below illustrate my findings with several freely available large datasets of molecules.
PAINS script speed tests
Jonathan B. Baell and Georgina A. Holloway published a very interesting paper on their analysis of frequent hitters from screening assays. DOI, more recently Walters et al DOI proposed and additional set of PAINS based on observations from a sulfhydryl-scavenging high-throughput screen. These structural motif definitions were combined into a single script that now contains a total of 487 SMARTS definitions. The table below shows the time taken to annotate several databases using the modified script. It should be noted that the filters cannot be truly comprehensive since they can only test the chemical space encompassed by the original compound collection. As shown by the Walters paper additional PAINS will certainly be identified as novel chemical space is explored. The table below shows the times for the script to run on several datasets of varying sizes, times are in hours:mins:seconds format.
UPDATE. There have now been a couple of new publications describing the identification of false positives in high-throughput screening campaigns in which the binding of glutathione S-transferase (GST) to glutathione (GSH) is used for detection of GST-tagged proteins.
Identification of Small-Molecule Frequent Hitters of Glutathione S-Transferase–Glutathione Interaction [DOI]
Identification of Small-Molecule Frequent Hitters from AlphaScreen High-Throughput Screens [DOI]
There have also been some suggestions as to how some of the motifs might be interfering with the assay.
I’ve now added the additional structural motif definitions taking the total to 550 SMARTS definitions. It is perhaps worth mentioning that some of these motifs may not be an issue when using alternative screening technologies, but it may be very worthwhile to double check any molecules flagged by this script before committing significant resources to follow up.
IncCalc SMILES refers to the total time taken including calculating and indexing SMILES, PreCalc SMILES refers to the search using pre-calculated SMILES. NT is Not Tested
| Database | Number of Molecules | IncCalc SMILES | PreCalc SMILES |
|---|---|---|---|
| ChEMBL_20 | 1,456,020 | 6:10 | 5:10 |
| ZINC | 22,723,296 | 1:43:20 | 55:10 |
| PubChem* | 67,441,980 | NT | 3:50:20 |
It is worth highlighting the speed of this pattern matching, there are 487 SMARTS queries in the filter which means the process is running at 2.36M matches per second!
Generating SMILES
To achieve the speed in searching indexed SMILES are used, this means that even if you open a file in SDF format a column containing the SMILES will need to be created. The first part of the script creates a column containing a SMILES string representation of the molecule, for larger datasets it may be actually be faster to first calculate the SMILES using the Tools menu. For the ZINC dataset of just over 22M molecules generating the SMILES from the Tools menu took just over 30 mins compared to approaching an hour using the script.
Another point worth noting for these large files is that even if you open a compressed SDF file (filename.sdf.gz) the file is first uncompressed and stored in a temp folder, Vortex then just keeps the byte wise seek into the original file then fetches MOL files on demand from disk. Disk space can thus appear to disappear! Importing SMILES is probably a better option in the longer term.
Dundee Filter
University of Dundee NTD Screening Library Filters ChemMedChem. 3(3): 435444. Compounds containing unwanted functionalities were removed as it is not desirable to waste resources removing such functionalities in the hit list. These included potentially mutagenic groups such as nitro groups, groups likely to have unfavourable pharmacokinetic properties such as sulphates and phosphates; and reactive groups such as 2-halopyridines or thiols.
This script contains 101 SMARTS definitions
| Database | Number of Mols | IncCalc SMILES | PreCalc SMILES |
|---|---|---|---|
| ChEMBL_20 | 1,456,020 | 5:48 | 4:50 |
| ZINC | 22,723,296 | 1:45:20 | 58:20 |
| PubChem | 67,441,980 | NT | 3:35:20 |
Liver toxicity Filter
Wallqvist et al have identified 12 molecular fragments that are strongly associated with drug-induced human liver injuries. DOI using data from LiverTox a database of information on the diagnosis, cause, frequency, patterns, and management of liver injury attributable to prescription and nonprescription medications, herbals and dietary supplements.
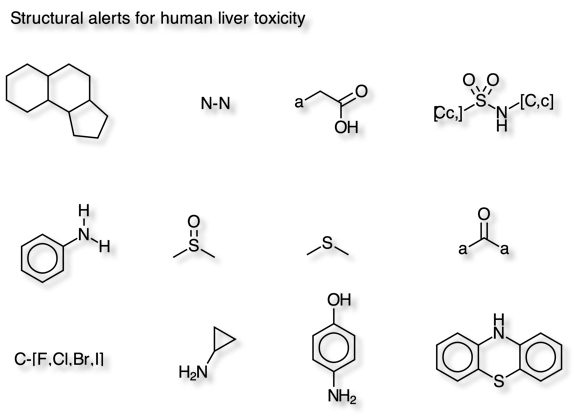
The script was tested using the dataset provided in the supplementary information.
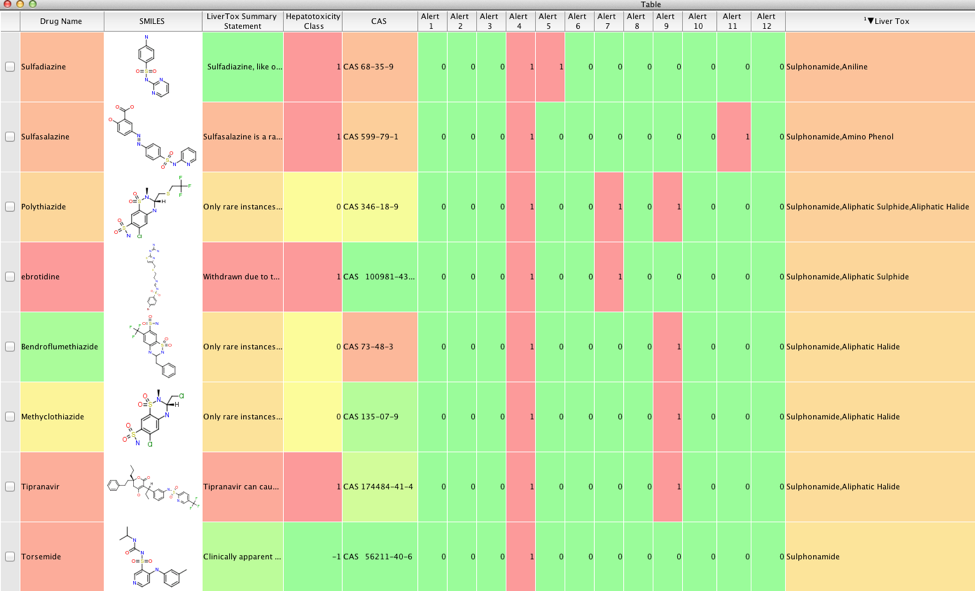
| atabase | Number of Molecules | IncCalc SMILES | PreCalc SMILES |
|---|---|---|---|
| ChEMBL_20 | 1,456,020 | 1:27 | 0:34 |
| ZINC | 22,723,296 | 44:37 | 9:27 |
| PubChem | 67,441,980 | NT | 22:26 |
Databases
ChEMBL is a database of bioactive drug-like small molecules, it contains 2-D structures, calculated properties (e.g. logP, Molecular Weight, Lipinski Parameters, etc.) and abstracted bioactivities (e.g. binding constants, pharmacology and ADMET data).
ZINC is a free database of commercially available compounds for virtual screening. Irwin, Sterling, Mysinger, Bolstad and Coleman, J. Chem. Inf. Model. 2012 DOI
PubChem, released in 2004, provides information on the biological activities of small molecules.
*Using PubChem
Whilst importing ChEMBL and ZINC proceeded uneventfully PubChem presented a few problems. I downloaded PubChem as a series of compressed sdf files, these I then combined into a single file. This generated a 55 GB sdf.gz file, reading this into Vortex proved problematic and I suspect there are a number of poorly defined records. So I converted it to SMILES using the following python script provided by Matt.
#!/usr/bin/env python
# -*- coding: utf-8 -*-
"""
pubchem2smi.py
~~~~~~~~~~~~~~
Read PubChem SDF files and output SMILES for each molecule.
Usage
python pubchem2smi.py path/to/Compound_*.sdf.gz > smiles.txt 2> log.txt
"""
from __future__ import print_function
from __future__ import unicode_literals
from __future__ import division
import gzip
import sys
from rdkit import Chem
def readSdf(filename):
print('%s\nReading file: %s\n%s' % ('='*100, filename, '-'*100), file=sys.stderr)
success, fail = 0, 0
for rdmol in Chem.ForwardSDMolSupplier(gzip.open(filename)):
if rdmol is None:
print(' => Failed to read molecule', file=sys.stderr)
fail += 1
continue
try:
smiles = Chem.MolToSmiles(rdmol, isomericSmiles=True)
except (ValueError, RuntimeError):
print(' => Failed to generate SMILES', file=sys.stderr)
fail += 1
continue
success += 1
print('%s\t%s' % (smiles, rdmol.GetProp('PUBCHEM_COMPOUND_CID'.encode())))
print('%s\nSummary for: %s' % ('-'*100, filename), file=sys.stderr)
print(' => %s successes, %s failures\n%s\n\n\n' % (success, fail, '='*100), file=sys.stderr)
if __name__ == '__main__':
if len(sys.argv) < 2:
print('Usage: %s file.sdf.gz' % sys.argv[0], file=sys.stderr)
sys.exit(1)
for filename in sys.argv[1:]:
readSdf(filename)
This yielded a file containing 67,442,980 SMILES (suggesting many structures were not converted), this I imported into Vortex, when I tried to run the scripts they hung when indexing the SMILES. Suspecting there might be problematic SMILES I used Vortex to recalculate the SMILES strings and then deleted the column containing the RDKit SMILES. At this point Vortex was using 48GB of RAM so I saved the file as vortex.gz and restated the computer to start with a clean sheet. I then reopened the file and ran a substructure search to force indexing of the SMILES strings, this worked without a hitch. I then ran the PAINS script which took nearly 4 hours but did annotate over 67 million structures.
Summary
I think it is pretty impressive that a desktop application like Vortex can handle such huge datasets, I suspect I’m pretty much on the limit for my machine (Mac Pro, 3.5 GHz 6-Core Intel Xeon E5, 64 GB RAM) but since PubChem is probably the largest publicly available compound collection I’m not too worried.
The Vortex scripts can be downloaded from here.
Page Updated 16 August 2017
2 thoughts on “Vortex script for Substructure searching very large compound collections.”
Comments are closed.