PROteolysis TArgeting Chimeras (PROTACs) technology provides an alternative to module biological function by specially using the ubiquitin proteasome system to induce degradation of the target protein DOI.
The PROTAC is composed of three components.
- A head-group that targets the protein of interest
- A crosslinker
- A second ligand at the opposite end that binds an E3 ligase
PROTAC ligand binds to both the protein target and an E3 ubiquitin ligase to form a ternary complex, followed by transfer of ubiquitin from the E2 to the protein substrate. The ubiquitin is attached to a lysine on the target protein, subsequent ubiquitins are then added to a lysine residue of the first added ubiquitin. The ternary complex dissociates and PROTAC is recycled. Polyubiquitinated protein undergoes degradation via 26S proteasomes.
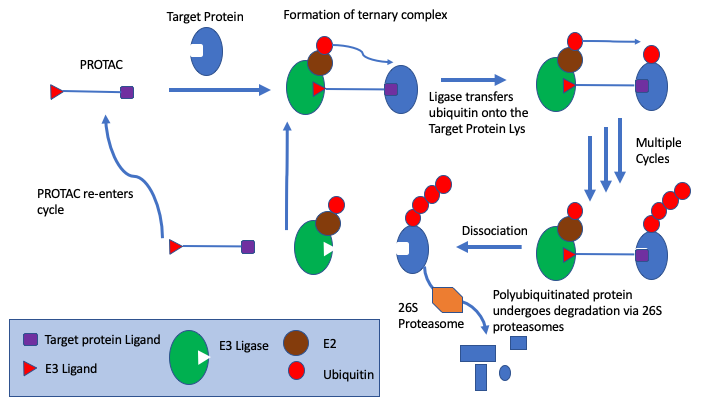
There are now many E3 ligases identified and multiple ligands for each ligase, a wide variety of cross linker have been employed and of course the head-group will vary depending on the protein of interest (POI). Whilst the PROTACs have a similar modular structure annotation of the different substructures is a challenge. In the past I’ve used a series of SMARTS queries but this becomes difficult to maintain, especially when one SMARTS string can be a substring of a larger SMARTS query.
Recently Astra Zeneca published a machine learning framework to split PROTACs into the different components. The code is freely available on GitHub.
https://github.com/ribesstefano/PROTAC-Splitter
I created a conda environment and then installed the package, note it is only tested using python 3.10.8. I also installed Jupyter
|
1 2 3 4 5 |
conda create --name protacSplit python=3.10.8 pip install git+https://github.com/ribesstefano/PROTAC-Splitter.git conda install Jupyter |
I then created a Jupyter notebook as shown below.
import pandas as pd
from protac_splitter import split_protac
protac_smiles = "CC(C)(C)S(=O)(=O)c1cc2c(Nc3ccc4scnc4c3)ccnc2cc1OCCOCCOCCOCCOCC(=O)Nc1cccc2c1CN(C1CCC(=O)NC1=O)C2=O"
ligands = split_protac(protac_smiles)
print(ligands)
Splitting PROTAC SMILES using XGBoost model: 0%| | 0/1 [00:00<?, ? examples/s]
{'text': 'CC(C)(C)S(=O)(=O)c1cc2c(Nc3ccc4scnc4c3)ccnc2cc1OCCOCCOCCOCCOCC(=O)Nc1cccc2c1CN(C1CCC(=O)NC1=O)C2=O', 'default_pred_n0': 'O=C1CCC(N2Cc3c(N[*:2])cccc3C2=O)C(=O)N1.O=C(COCCOCCOCCOCCO[*:1])[*:2].CC(C)(C)S(=O)(=O)c1cc2c(Nc3ccc4scnc4c3)ccnc2cc1[*:1]', 'model_name': 'XGBoost'}
from rdkit import Chem
from rdkit.Chem import Draw
from rdkit.Chem.Draw import IPythonConsole
from rdkit.Chem import PandasTools
from protac_splitter import split_protac
from protac_splitter import split_prediction
from protac_splitter import split_protac_graph_based
import numpy as np
import pandas as pd
df = pd.read_csv('/Users/chrisswain/Projects/Protacs/ForSplitter.csv')
print(df.shape)
(9161, 2)
df.head(5)
| InChI Key | SMILES | |
|---|---|---|
| 0 | RPMQBLMPGMFXLD-PDUNVWSESA-N | COc1cc(cc(c1CN1CCN(CCOCCOCC(=O)N[C@H](C(=O)N2C... |
| 1 | RPMQBLMPGMFXLD-PDUNVWSESA-N | COc1cc(cc(c1CN1CCN(CCOCCOCC(=O)N[C@H](C(=O)N2C... |
| 2 | DINLAPHIPWQULR-VQNWZXTLSA-N | CCN(CCOCCOCC(=O)N[C@H](C(=O)N1C[C@H](O)C[C@H]1... |
| 3 | YPKQEFMVVKVSQU-RXJDTIOWSA-N | Cc1ncsc1-c1ccc(cc1)CNC(=O)[C@@H]1C[C@@H](O)CN1... |
| 4 | ZSCOIFSUFMYZEZ-YSWDPXALSA-N | Cc1ncsc1-c1ccc(cc1)CNC(=O)[C@@H]1C[C@@H](O)CN1... |
Split_df = split_protac(df, protac_smiles_col='SMILES')
PandasTools.AddMoleculeColumnToFrame(Split_df, smilesCol='default_pred_n0')
Splitting PROTAC SMILES using XGBoost model: 0%| | 0/9161 [00:00<?, ? examples/s]
Split_df.head(5)
| SMILES | default_pred_n0 | model_name | ROMol | |
|---|---|---|---|---|
| 0 | COc1cc(-c2cn(C)c(=O)c3cnccc23)cc(OC)c1CN1CCN(C... | Cc1ncsc1-c1ccc(CNC(=O)[C@@H]2C[C@@H](O)CN2C(=O... | XGBoost | <rdkit.Chem.rdchem.Mol object at 0x36d7ebb50> |
| 1 | COc1cc(-c2cn(C)c(=O)c3cnccc23)cc(OC)c1CN1CCN(C... | Cc1ncsc1-c1ccc(CNC(=O)[C@@H]2C[C@@H](O)CN2C(=O... | XGBoost | <rdkit.Chem.rdchem.Mol object at 0x36d7eb840> |
| 2 | CCN(CCOCCOCC(=O)N[C@H](C(=O)N1C[C@H](O)C[C@H]1... | Cc1ncsc1-c1ccc([C@H](C)NC(=O)[C@@H]2C[C@@H](O)... | XGBoost | <rdkit.Chem.rdchem.Mol object at 0x36d7eb990> |
| 3 | Cc1ncsc1-c1ccc(CNC(=O)[C@@H]2C[C@@H](O)CN2C(=O... | Cc1ncsc1-c1ccc(CNC(=O)[C@@H]2C[C@@H](O)CN2C(=O... | XGBoost | <rdkit.Chem.rdchem.Mol object at 0x36d7eba00> |
| 4 | Cc1ncsc1-c1ccc(CNC(=O)[C@@H]2C[C@@H](O)CN2C(=O... | Cc1ncsc1-c1ccc(CNC(=O)[C@@H]2C[C@@H](O)CN2C(=O... | XGBoost | <rdkit.Chem.rdchem.Mol object at 0x36d7eb060> |
Split_df.to_csv('output.csv', columns=['default_pred_n0'],index=False, header=False)
The cell below splits a single PROTAC and the output shows the format of the result. The individual components of the PROTAC are output as SMILES string separated by a full stop (period).
|
1 2 3 4 5 |
protac_smiles = "CC(C)(C)S(=O)(=O)c1cc2c(Nc3ccc4scnc4c3)ccnc2cc1OCCOCCOCCOCCOCC(=O)Nc1cccc2c1CN(C1CCC(=O)NC1=O)C2=O" ligands = split_protac(protac_smiles) print(ligands) {'text': 'CC(C)(C)S(=O)(=O)c1cc2c(Nc3ccc4scnc4c3)ccnc2cc1OCCOCCOCCOCCOCC(=O)Nc1cccc2c1CN(C1CCC(=O)NC1=O)C2=O', 'default_pred_n0': 'O=C1CCC(N2Cc3c(N[*:2])cccc3C2=O)C(=O)N1.O=C(COCCOCCOCCOCCO[*:1])[*:2].CC(C)(C)S(=O)(=O)c1cc2c(Nc3ccc4scnc4c3)ccnc2cc1[*:1]', 'model_name': 'XGBoost'} |
The rest of the notebook imports a file containing 9000 PROTACs into a pandas data frame and then splits them into the individual components, the results are then exported to a text file "output.csv", removing the index and header.
The result is a file containing the three components separated by a full stop, with linking positions annotated.
O=C1CCC(N2C(=O)c3cccc(N[:2])c3C2=O)C(=O)N1.O=C(COCCOCC[:1])[:2].O=c1c2nc3ccccc3[nH]c-2nn1[:1]
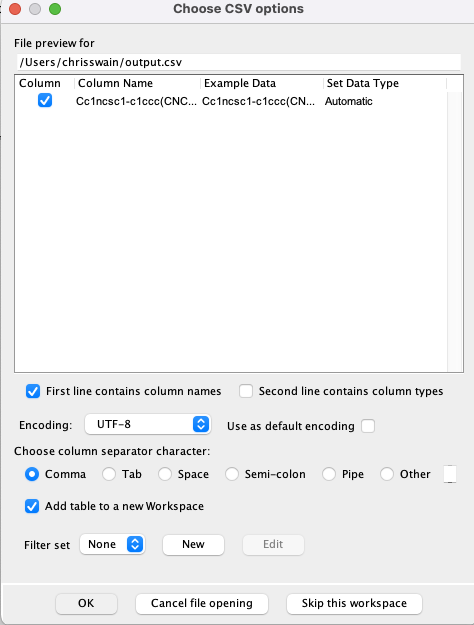
We can now import this file into Vortex, as you can see in the image below the default is to import as a comma separated values. However in this case we need to use a full stop as the separator.
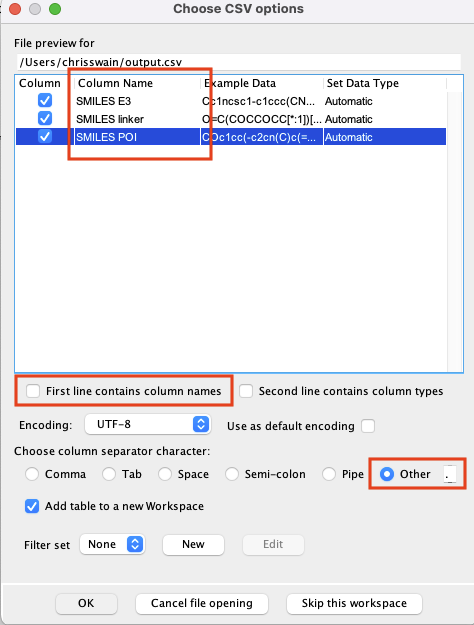
We can do this by modifying the import options as shown below. First unselect "First line contains column names", then choose "other" as the column separator character, and type a full stop (period) into box. The file preview should now update to have column names (the default is C0,C1,C2) you now need to type in the names as shown below.
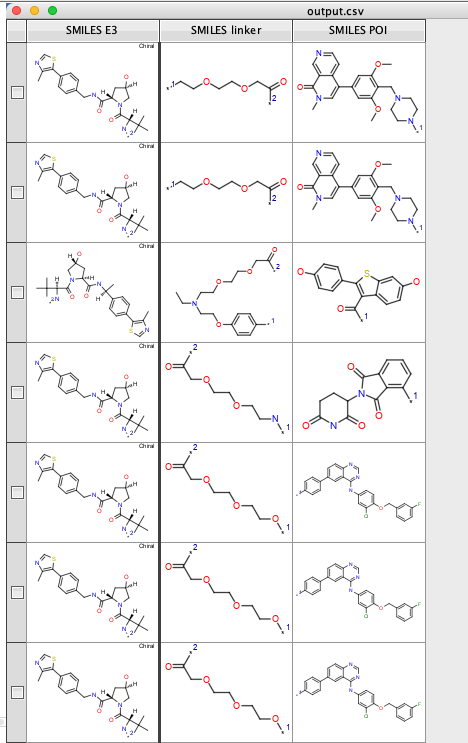
Click "OK" and the file will be imported.
You can then use the "Generic Category script" from the cluster analysis collection to see how any of each E3 ligase ligand are employed https://macinchem.org/2023/03/11/a-collection-of-vortex-scripts-to-aid-cluster-analysis/
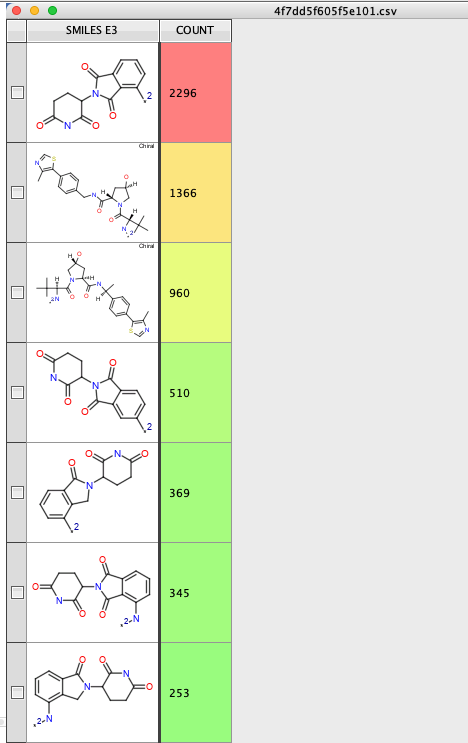
Many thanks to AZ for making this tool available to the community.