Rowan is a software platform that makes it possible to run high-level quantum chemical calculations through a web interface. In general, running quantum chemical calculations (like DFT or Hartree–Fock) is difficult for a few reasons: the calculations are very computationally expensive and take hours or days to run, so you need to run them on an external compute cluster, and there’s no GUI so a good amount of wrangling on the command line is needed to submit, monitor, and analyze jobs. As a result, most researchers who aren’t hardcore computational chemists tend to avoid submitting quantum chemical calculations, even though the results for e.g. property prediction or molecular geometries are often much more accurate than the alternatives.
Rowan aims to fix these issues and lower the barrier to entry by providing an integrated web platform for computational chemistry. You can upload data in a variety of formats to Rowan (sdf, mol2, xyz, mae, SMILES), select the calculations that you want to be performed, and then submit the jobs – Rowan automatically allocates cloud computing resources, runs the jobs, and displays the results as the calculations run. There’s no upfront cost to use Rowan, you just get billed for the computer time that your calculations use.
Recently machine-learned interatomic potentials have attracted attention as low-cost replacements for conventional quantum chemistry DOI – ML potentials trained on DFT energies and forces combine the speed of forcefields with the accuracy of quantum chemistry. You can run the brand-new AIMNet2 machine-learned interatomic potential on Rowan, which gives generally excellent results for druglike molecules while running two to three orders of magnitude faster than DFT calculations of similar accuracy.
Here’s what submitting a dihedral scan on rivaroxaban through Rowan looks like. The input structure was automatically generated from a SMILES string.
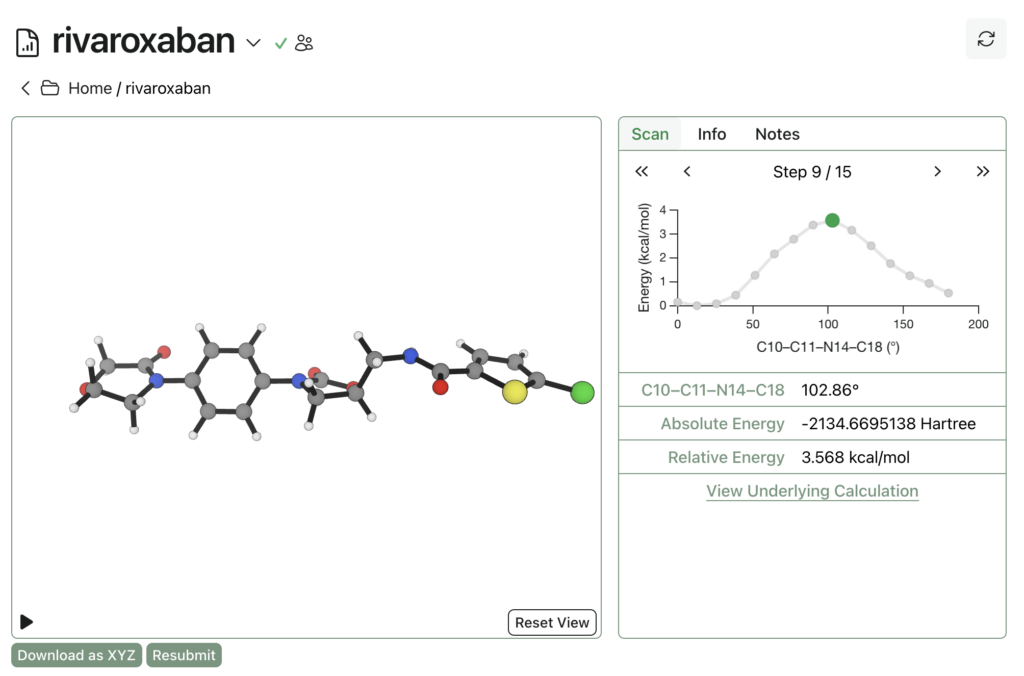
Rowan uses the wavefront propagation method to generate smooth and accurate scan surfaces without artifacts from imperfect relaxation. The entire relaxed dihedral scan spawns 48 individual calculations, cost 164 credits (about $3), and took under an hour to complete: here’s a link to the output, and here’s what it looks like:
Calculations run on Rowan are private by default, but can be shared publicly if desired. Support for organizations and sharing with individuals is planned but doesn’t exist yet. There’s also a Google Drive-like way to group calculations into folders through Rowan, and ways to add notes to calculations and folders to stay organized.
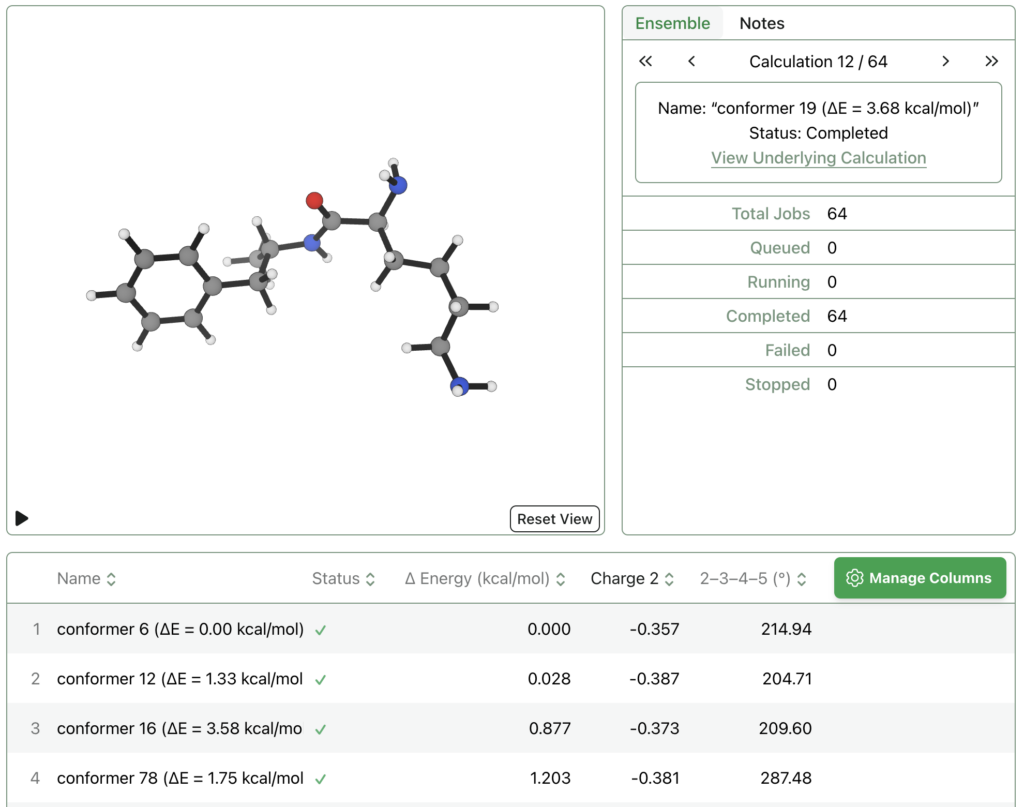
Rowan also allows for calculations to be run on lots of structures at once through “Ensembles.” You can upload a list of different conformers from RDKit, for instance, and then optimize each of them and compare the final energies, bond distances, or other properties. Rowan runs all the jobs in parallel so it only takes a few minutes.
The image below shows 64 different conformers of lisdexamfetamine that were optimized and sorted by relative energy – this data can also be exported as a csv file for easy analysis. The molecule names show the MMFF relative energies; there are plenty of low-lying conformers which MMFF erroneously predicts to be high in energy, demonstrating how quantum chemistry can be helpful.
Rowan is still very new—the company is only seven months old—but has potential to be a compelling alternative to conventional programs for plenty of use cases. The interface is simple enough that it’s possible to use even without any previous experience with quantum chemistry, and even the current feature set will already be useful in many different workflows. You get 500 free credits when you sign up, so it’s worth checking out! You can make an account with Rowan here.
Alternatively, you can use the Rowan python api (you will need an API key).
|
1 |
pip install rowan-python. |
Details are on GitHub https://github.com/rowansci/rowan-python