The 2015.10 release of Chemical Computing Group’s Molecular Operating Environment (MOE) software includes a number of new features, enhancements and changes and I thought it might be useful to have a more detailed look at the update.
One of the more interesting feature is the move towards remote computation. A number of applications can now off-load heavy-duty calculations to a remote server, this offers the possibility of performing the computations on a cluster or even in the cloud. You will of course need the appropriate MOE licenses to run on multiple cores.
The following applications have been remote server-enabled:
Pharmacophore Search
Project Search
PDB Search
Domain Motif Search
Loop Modeler
PLIF (Protein Ligand Interaction Fingerprints)
Cloud service providers such as Amazon Web Services (AWS) provide rapidly scalable on-demand compute and storage capacity allowing large scale, parallelisable computations to be carried out quickly and in real-time. The MOE help provides a detailed explanation of all the steps necessary to launch an AWS cluster running MOE, all communications between the cluster will be via encrypted provided that SSH is used.
I thought it might be interesting to see how the new Pharmacophore Search performs on my MacPro since it has 12 cores. Ideally you want use this with really big databases, and CCG provide a version of ChEMBL20from their downloads. To do this we first need to split the large file into multiple chunks, one for each core that you want to use. CCG provide a handy SVL script for doing this dbsplit.svl. Simply load the svl script and then in the SVL command window type.
svl> db_split ['chemblr20_frags',100000]
This gives us a folder containing 9 databases, these now need to be put into a specific folder so that the moe web server can find them. They need to be in this specific directory
$HOME/moefiles/moeweb/perm/shared/ph4
You might need to create the ph4 directory in shared as it wasn’t present in my installation.
Starting up the MOE web server
We can then start up the moe web server
MacPro:~ Chris$ /Applications/moe2015/bin/moeweb -work 11 — serverName: macpro.home — serverHost: macpro.home — serverIP: 192.148.1.112 — init-rc: /Users/Chris/moefiles/init-rc (not found) — web-rc: /Users/Chris/moefiles/web-rc (not found) — RC host: macpro.home — RC arch: mac64 — RC os: unix — nhosts: 11
If you now start up the MOE GUI and click on the cog icon in the top right of the interface and then select MOE/web Settings from the dropdown menu.
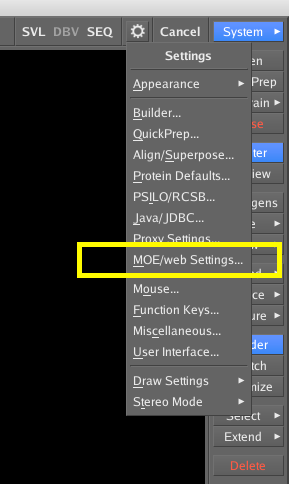
Hopefully you should see everything in green as shown in the image below. One of the things that can be a potential issue is if you have a local firewall running, in which case you might need to change the settings to allow access on port 8888.
Running a Pharmacophore Search
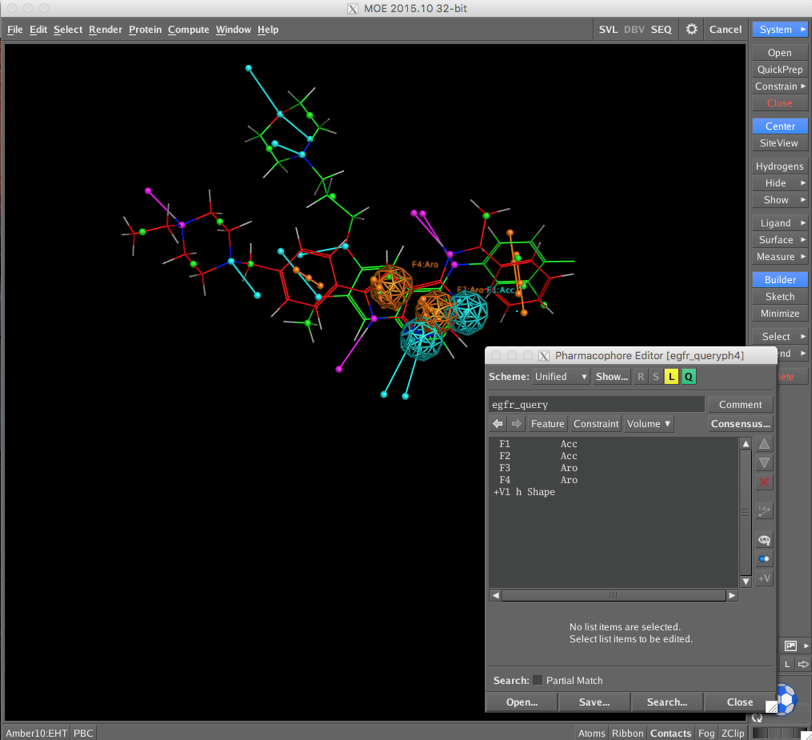
CCG provide a sample pharmacophore query (/Applications/moe2015/sample/mol/egfr_query.ph4) which is shown below superimposed on a couple of known ligands. The Pharmacophore Editor allows you add or amend the pharmacophoric features but I just used the query as provided.
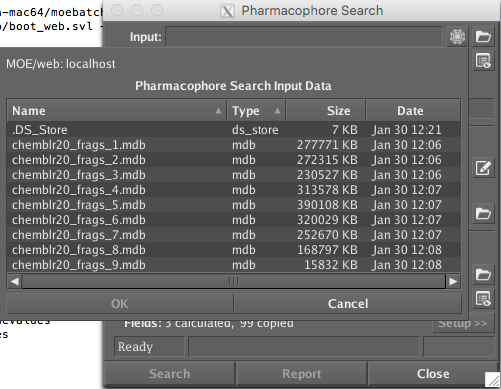
At the moment crashes after 85% completion!!
pKa Prediction
One of the new features is the ability to add a set of Hueckel Theory descriptors reports quantities based on averages over all protonation states at pH 7 pKa as descriptors in the database viewer, including pKa.
- h_pavgQ: Average total charge sum.
- h_pstates: Entropic count (fractional number of protonation states).
- h_pstrain: Strain energy to recover input protonation state.
- h_pKa: pKa of the reaction to remove a proton.
- h_pKb: pKb of the reaction to add a proton.
- h_logD: logD as a state average.
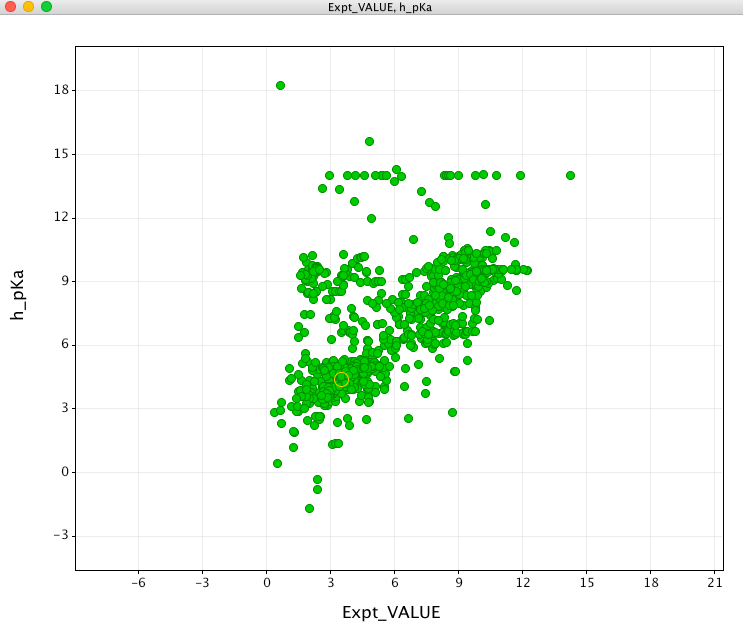
I have a database of experimental pKa values that I used to evaluate the pKa prediction. First looking at the prediction for molecules containing an acidic group (shown in green in the plot below). In general there is a reasonable correlation.
However when looking at the prediction for molecules containing basic sites the results are rather perplexing, as shown in yellow below. If anything there appears to be an inverse correlation?
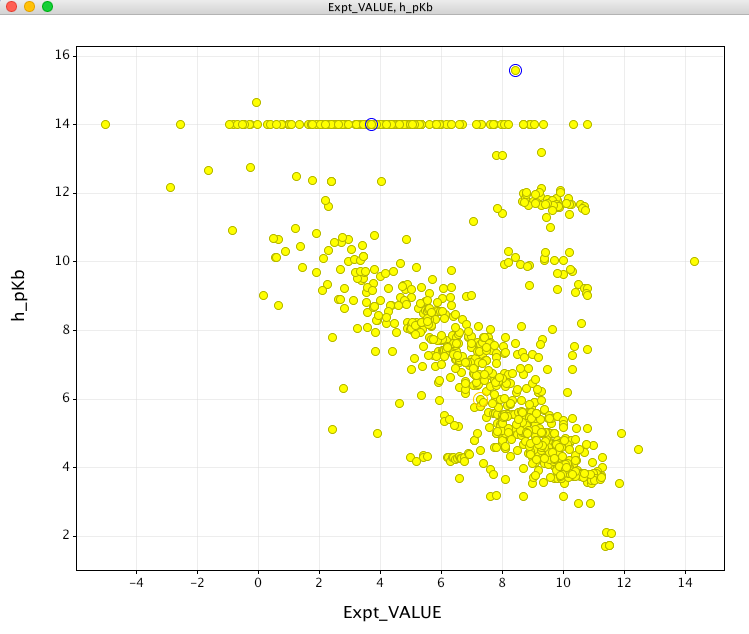
After chatting with the support at CCG it seems..
It turns out that the way we do our calculation you need to calculate 14 minus the value of the pKb descriptor
If I then subtract the calculated result from 14 I get the plot below, this is obviously an improvement, inspecting some of the outliers (blue circles) it seems that these are all molecules that would be predicted to be zwitterions.
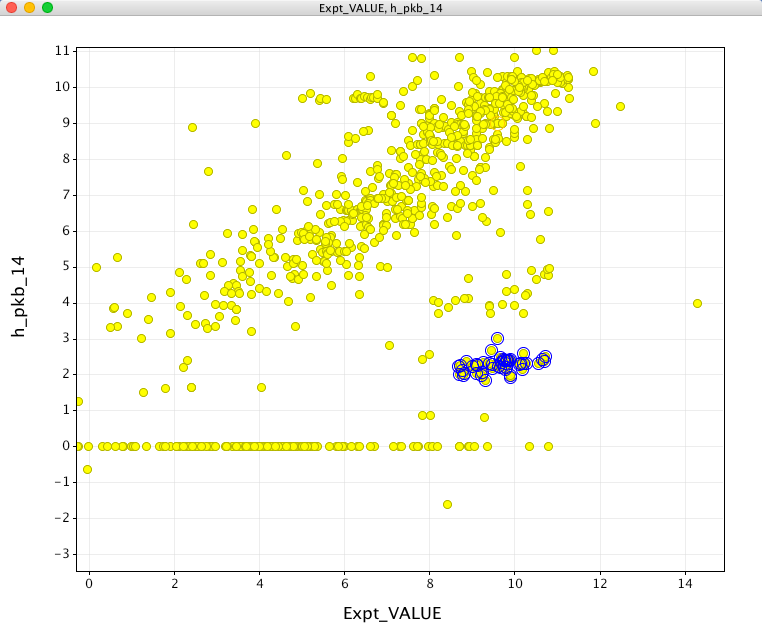
Overall it looks like it will be useful but I suspect most users would not expect to have to have to subtract from 14 to bring the calculated results onto the expected scale. The calculation also returns a value of 14 if no ionisable sites are found, personally I would have preferred it to simply return no result.
Docking
Docking has had a major update
Covalent docking has now been implemmented. Reaction-based covalent docking is now available through the Dock panel, it uses a reaction transform to determine how the ligand is bonded to the receptor.
If you are going to add your own reactions it is worth having a look at the examples provided. The reactions are read in from RDF files and are located in $MOE/lib/reactions/covalent. The image below shows the front for the reaction of a nucleophilic Ser, Thr or Cys with a Micheal acceptor.
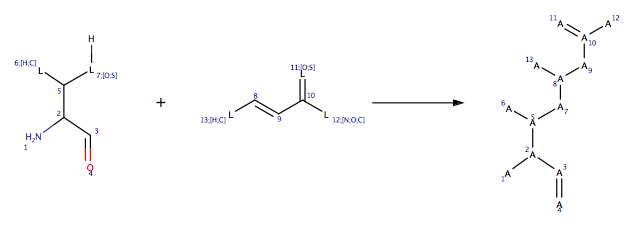
The interface provides the possibility of specifying a Reactive Site, which is a subset of the receptor atoms, and can be used to target individual residue(s) or even atoms.
Other Features
Some other features caught my eye. Some time ago I wrote and article on 3D stereo viewing on a Mac in particular the use of the new 3D TV, this has received extra support in the latest MOE update.
3D TV and 3D SpaceMouse support. MOE now supports 3D TV side-by-side, over-under, and DLP stereo modes. SpaceMouse 6DOF devices from 3Dconnexion are also now supported.
Whilst I have a couple of tools that can predict NMR spectra it is not possible to do much more than do a side by side comparison by eye.
NMR Analysis. The new DBV | Compute | Molecule | NMR Analysis application, allows for the comparison of experimental to calculated 13C NMR chemical shifts, for determining and characterizing chemical structures.
Last Updated 3 February 2016