Small molecules can potentially bind to a variety of bimolecular targets and whilst counter-screening against a wide variety of targets is feasible it can be rather expensive and probably only realistic for when a compound has been identified as of particular interest. For this reason there is considerable interest in building computational models to predict potential interactions. With the advent of large data sets of well annotated biological activity such as ChEMBL and BindingDB this has become possible.
ChEMBL 24 contains 15,207,914 activity data on 12,091 targets, 2,275,906 compounds, BindingDB contains 1,454,892 binding data, for 7,082 protein targets and 652,068 small molecules.
These predictions may aid understanding of molecular mechanisms underlying the molecules bioactivity and predicting potential side effects or cross-reactivity.
Whilst there are a number of sites that can be used to predict bioactivity data I’m going to compare one site, Polypharmacology Browser 2 (PPB2) http://ppb2.gdb.tools with two tools that can be downloaded to run the predictions locally. One based on Jupyter notebooks models built using ChEMBL built by the ChEMBL group https://github.com/madgpap/notebooks/blob/master/targetpred21_demo.ipynb and a more recent random forest model PIDGIN. If you are using proprietary molecules it is unwise to use the online tools.
PPB2
PPB2 was built from bioactivity dataset of all compounds having at least IC50 < 10 uM on a single protein target in ChEMBL22, considering only high confidence data points as annotated in ChEMBL and only targets for which at least 10 compounds were documented. This provided 344,163 single compounds associated with 1,720 single protein targets belonging to 8 different target families, representing 555,346 target-compound associations.
To encode molecular structures we selected three fingerprints perceiving different levels of details, namely: 1) MQN(Molecular Quantum Number), a 42-bit scalar fingerprint representing molecular composition by atoms, bonds, polar groups and topological features36, 37particularly useful to search and visualize large databases;38-402)Xfp (atom category extended atom-pair fingerprint), an 55-bit scalar fingerprint perceiving molecular shape and pharmacophores and well suited for scaffold-hopping virtual screening;41and 3)ECfp4(extended connectivity fingerprint up to four bonds), a 1024-bit binary substructure fingerprint encoding detailed information about molecular structure.
Several different searches are possible
Nearest neighbour search with:
- Extended Connectivity fingerprint ECfp4 NN(ECfp4)
- Shape and Pharmacophore fingerprint Xfp NN(Xfp)
- Molecular Quantum Numbers MQN NN(MQN)
ECfp4 Naive Bayes Machine Learning model produced on the fly with 2000 nearest neighbors from:
- Extended Connectivity fingerprint ECfp4 NN(ECfp4) + NB(ECfp4)
- Shape and Pharmacophore fingerprint Xfp NN(Xfp) + NB(ECfp4)
- Molecular Quantum Numbers MQN NN(MQN) + NB(ECfp4)
Naive Bayes machine learning model with entire dataset using:
- Extended Connectivity fingerprint ECfp4 NB(ECfp4)
Deep Neural Network model with entire dataset using:
- Extended Connectivity fingerprint DNN(ECfp4)
Based on the published data the best performing models were, Extended Connectivity fingerprint ECfp4 NN(ECfp4), and Shape and Pharmacophore fingerprint Xfp NN(Xfp),
ChEMBL Model
Starts with a carefully selected subset of ChEMBL 24 data containing pairs of compounds and single-protein targets. They used two activity cut-offs, namely 1uM and a more relaxed 10uM, which correspond to two models trained on bioactivity data against 1028 and 1569 targets respectively. Morgan fingerprints (radius=2) were calculated using RDKit and then used to train a multinomial Naive Bayesian multi-category scikit-learn model. These models then were used to predict targets for the small molecule drugs. The Jupyter notebook is available for download.
PIDGIN
Bioactive molecules were extracted from ChEMBL_21 for pChEMBL activity values -Log(Ki/Kd/IC50/EC50) greater than or equal to ‘5’ (10 µm) in binding and functional assays. Percentage activation and inhibition data was also extracted when the ‘Activity Comment’ was declared ‘active’. The dataset contains 766 ,515 active data points, spanning 1,651 human targets. PubChem was mined for inactive compounds in the same procedure to resulting in the extraction of 3 630 485 inactives spanning 1 440 targets
Protein target prediction using Random Forests (RFs) trained on bioactivity data from PubChem (extracted 07/06/18) and ChEMBL (version 24), using the RDKit (2048bit Rdkit Extended Connectivity FingerPrints (ECFP)) and Scikit-learn, which employ a modification of the reliability-density neighbourhood Applicability Domain (AD) analysis by Aniceto DOI. This project is the successor to PIDGIN version 1 DOI and PIDGIN version 2 DOI. 10,446 models generated at four different cut-off’s: 100μM, 10μM, 1μM and 0.1μM for 7,075 biological targets.
Detailed installation instructions are available on GitHub https://pidginv3.readthedocs.io/en/latest/, and can be installed using CONDA
conda create -c keiserlab -c rdkit -c sdaxen --name pidgin3_env python=2.7 pip e3fp scikit-learn=0.19.0 pydot graphviz
Commandline options are fully detailed
MacPro:PIDGIN Chris$ python2 /Users/Chris/Projects/PIDGIN/PIDGINv3/predict.py -h
Usage: predict.py [options]
Options:
-h, --help show this help message and exit
-f FILE Input smiles or sdf file (required)
-d DELIM, --smiles_delim=DELIM
Input file (smiles) delimiter char (default: white
space ' ')
--smiles_column=SMICOL
Input file (smiles) delimiter column (default: 0)
--smiles_id_column=IDCOL
Input file (smiles) ID column (default: 1)
-o FILE Optional output prediction file name
-t, --transpose Transpose output (rows are compounds, columns are targets)
-n NCORES, --ncores=NCORES
No. cores (default: 1)
-b BIOACTIVITY, --bioactivity=BIOACTIVITY
Bioactivity threshold (can use multiple split by ','.
E.g. '100,10'
-p PROBA, --proba=PROBA
RF probability threshold (default: None)
--ad=AD Applicability Domain (AD) filter using percentile of weights [float]. Default: 90 (integer for percentile)
--known_flag Set known activities (annotate duplicates between input to train with correct label)
--orthologues Set to use orthologue bioactivity data in model
generation
--organism=ORGANISM Organism filter (multiple can be specified using commas ',')
--target_class=TARGETCLASS
Target classification filter
--min_size=MINSIZE Minimum number of actives used in model generation (default: 10)
--performance_filter=P_FILT
Comma-seperated performance filtering using following
nomenclature: validation_set[tsscv,l50so,l50po],metric
[bedroc,roc,prauc,brier],performance_threshold[float].
E.g 'tsscv,bedroc,0.5'
--se_filter Optional setting to restrict to models which do not require Sphere Exclusion (SE)
--training_log Optional setting to add training_details to the
prediction file (large increase in output file size)
--ntrees=NTREES Specify the minimum number of trees for warm-start
random forest models (N.B Potential large latency/memory cost)
--preprocess_off Turn off preprocessing using the flatkinson (eTox)
standardizer (github.com/flatkinson/standardiser),
size filter (100 >= Mw >= 1000 and organic mol check
(C count >= 1)
--std_dev Turn on matrix calculation for the standard deviation of prediction across the trees in the forest
--percentile Turn on matrix calculation for the percentile of AD compounds
--model_dir=MODEL_DIR
Path to directory containing models, if not default
MacPro:PIDGIN Chris$
I used—ncores=8, and I compared the default Applicability Domain —ad=90 (most stringent) with —ad=0 (no filter). PIDGINv3 applies the reliability-density neighbourhood Applicability Domain (AD) analysis by Aniceto et al., from: DOI.
Comparing the models
Comparing the models is actually non-trivial, it would be interesting to have a molecule with known target and off-target activities, however such molecules are likely to be in ChEMBL. Using recently published molecules means they won’t be in ChEMBL but unfortunately little off target info is likely to be published. I decided to use a selection molecules.
Clozapine
Clozapine is a well established neuroleptic drug, in a 2013 study DOI in a comparison of 15 antipsychotic drugs in effectiveness in treating schizophrenic symptoms, clozapine was ranked first and demonstrated very high effectiveness. Despite the widespread use the mode of action is still a little unclear and it has been shown to interact with a wide variety of receptors, ion channels and enzymes shown in the table below.
Values are Ki (nM). The smaller the value, the more strongly the drug binds to the site. All data are for human cloned proteins, except σ1 (guinea pig), MOR (rat), DOR (mouse), and KOR (guinea pig).
| Binding Site | CZP affinity |
|---|---|
| 5-HT1A | 123.7 |
| 5-HT1B | 519 |
| 5-HT1D | 1,356 |
| 5-HT2A | 5.35 |
| 5-HT2B | 8.37 |
| 5-HT2C | 9.44 |
| 5-HT3 | 241 |
| 5-HT5A | 3,857 |
| 5-HT6 | 13.49 |
| 5-HT7 | 17.95 |
| α1A | 1.62 |
| α1B | 7 |
| α2A | 37 |
| α2B | 26.5 |
| α2C | 6 |
| β1 | 5,000 |
| β2 | 1,650 |
| D1 | 266.25 |
| D2 | 157 |
| D3 | 269.08 |
| D4 | 26.36 |
| D5 | 255.33 |
| H1 | 1.13 |
| H2 | 153 |
| H3 | >10,000 |
| H4 | 665 |
| M1 | 6.17 |
| M2 | 36.67 |
| M3 | 19.25 |
| M4 | 15.33 |
| M5 | 15.5 |
| σ1 | 5,000 |
| MOR | 1,000 |
| DOR | 1,000 |
| KOR | 1,000 |
| SERT | 1,624 |
| NET | 3,168 |
| DAT | >10,000 |
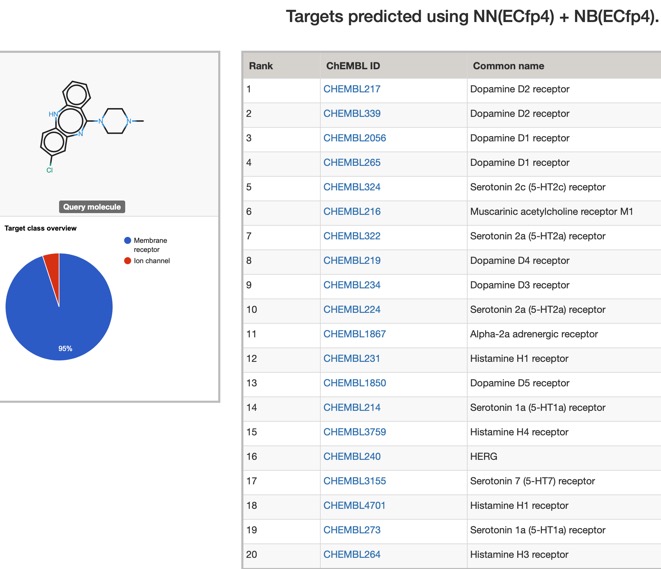
The predictions using PPB2 are shown in the image below
The predictions using the ChEMBL 10 uM model are shown below (only human displayed).
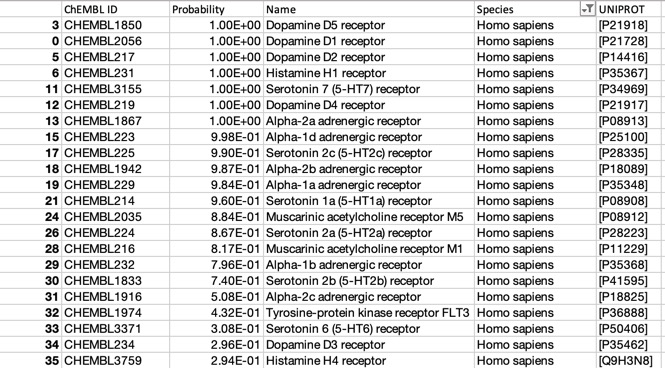
For such a promiscuous molecule the results from PPB2 and the ChEMBl models it is not surprising that both show broadly similar profiles with multiple bioactivities, with multiple GPCRs being flagged, PPB2 also flags the HERG ion channel and Clozapine is reported to have an affinity of 320 nM DOI.
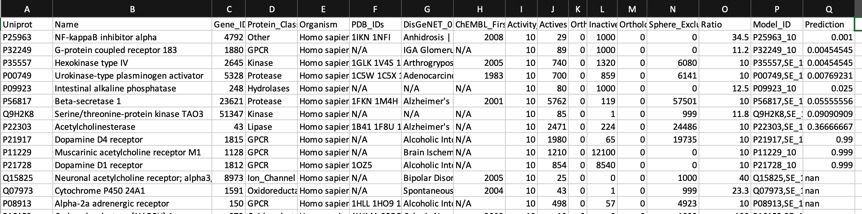
The predictions using the PIDGIN 10 uM model are shown below
Using default AD=90
Using AD=0
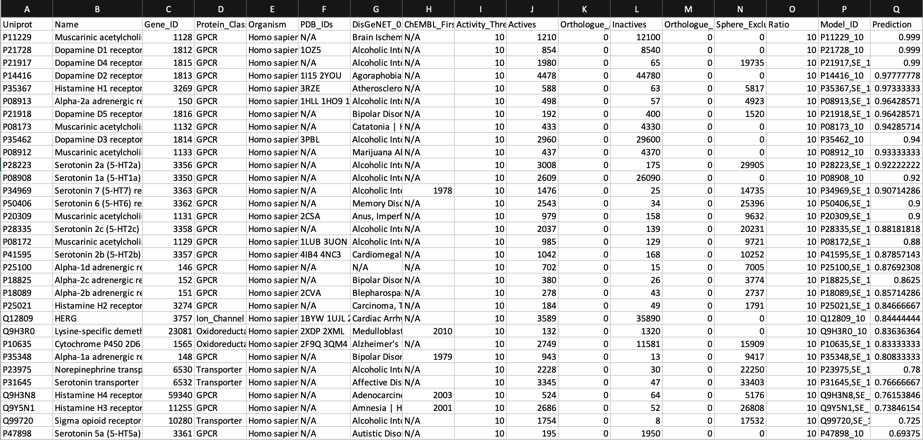
The default AD=90 flag of PIDGINv3 is quite stringent in assigning predictions in order to reduce the false positive rate, the AD=0 effectively turns this off. I suspect users will need to experiment a little with this parameter to get a feel for the most appropriate level.
One of the options for PIDGIN is
--known_flag
This annotates any known actives as shown below, it also flags any known inactives. For a compound like Clozapine this seems to be a useful since it lets you focus on potential bioactivities that have not been measured experimentally. As more data becomes available I can see this being useful, particularly for evaluating molecules from high-throughput screening.
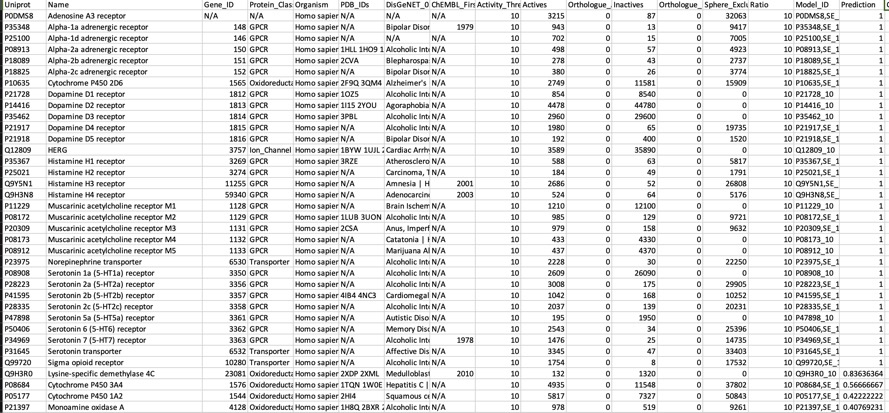
For a known promiscuous molecule like Clozapine all models correctly flag multiple activities for which known experimental data is available, perhaps unsurprisingly they also flag other activities for which there is no data in ChEMBL, the —known_flag in PIDGIN is very useful in this instance.
Bioactivity Prediction for a recently published molecule
A recent publication “Discovery of Indole- and Indazole-acylsulfonamides as Potent and Selective NaV1.7 Inhibitors for the Treatment of Pain” DOI describes 3-Aryl-indole and 3-aryl-indazole derivatives were identified as potent and selective Nav1.7 inhibitors. The reported activity is potent hNaV1.7 inhibition (IC50 = 108 nM) and good selectivity (hNaV1.5 IC50 =19 μM). Nav1.7 is a voltage-gated sodium channel and is also known as sodium voltage-gated channel alpha subunit IX, NaV1.5 is also known as sodium voltage-gated channel alpha subunit V. A structure search confirmed the molecule below was not yet in ChEMBL, and a similarity search within ChEMBL identified CHEMBL2323512 as the most similar compound (81%), this is reported to be an Aldo-keto reductase family 1 member C3 inhibitor DOI.
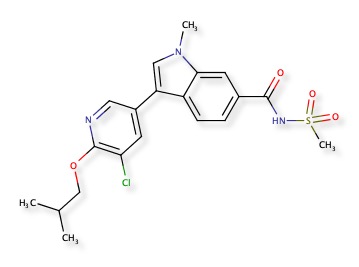
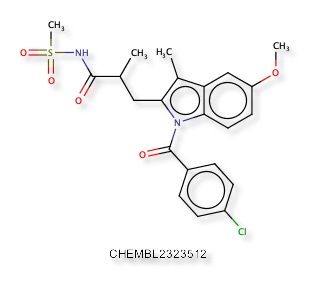
The predictions using PPB2 are shown in the image below.
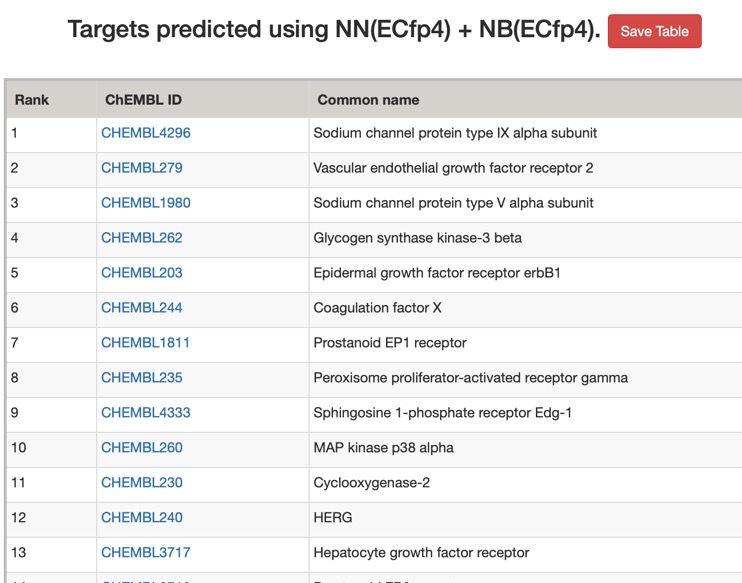
The predictions using the ChEMBL 10 uM model are shown below (only human displayed).
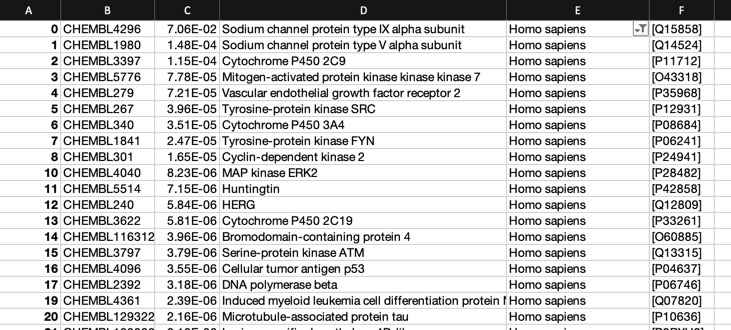
The predictions using the PIDGIN 10 uM model are shown below
Using default AD=90
Using AD=0
All models flag the sodium channel as a potential target, and also highlight kinases as potential off-target activities.
Bioactivity Prediction for a molecule from Open Source Malaria
OSM-S-106 is a screening hit from the Open Source Malaria project since this hit was identified using a phenotypic screen the mode of action is unknown, several exciting studies are underway to try and identify the mechanism but I thought it might be interesting to use these tools to try and predict potential biological targets. One issue in this sort of study is that the underlying data is heavily biased towards human targets. Never the less it might point towards targets that might be investigated in the malaria parasite, in addition knowledge of potential human off-target actives is also extremely useful since the eventual drug will be dosed to humans.
OSM-S-106 is actually in ChEMBL, CHEMBL581088 has very limited data in ChEMBL, mainly high-throughput screening information, and does not appear to have been cross-screened.
The predictions using PPB2 are shown in the image below.
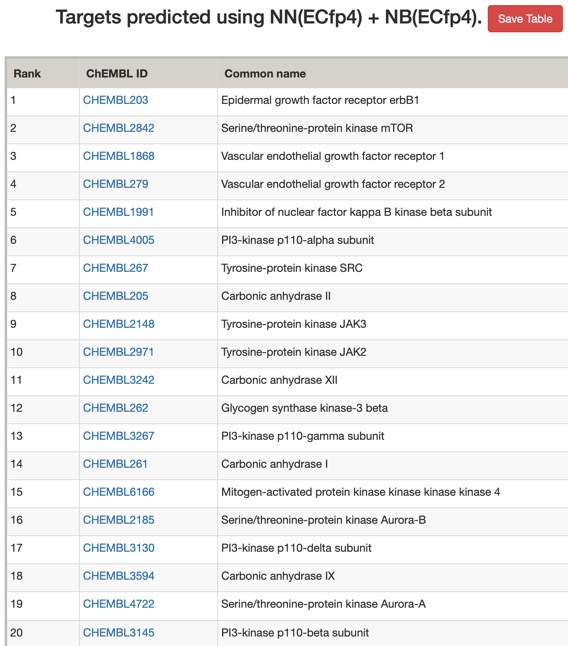
The predictions using the ChEMBL 10 uM model are shown below (all species displayed).
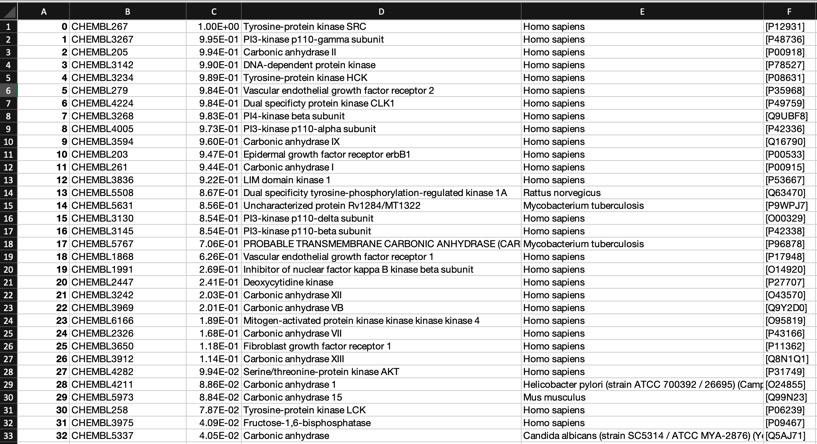
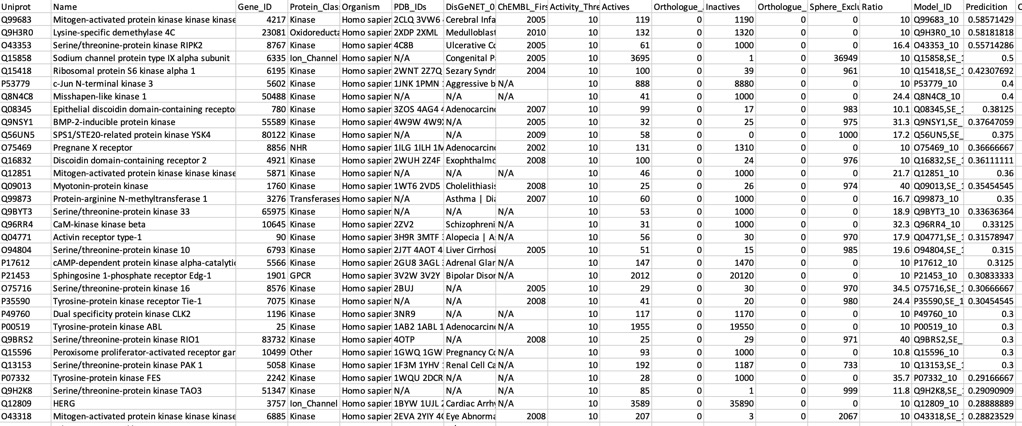
The predictions using the PIDGIN 10 uM model are shown below
Using default AD=90
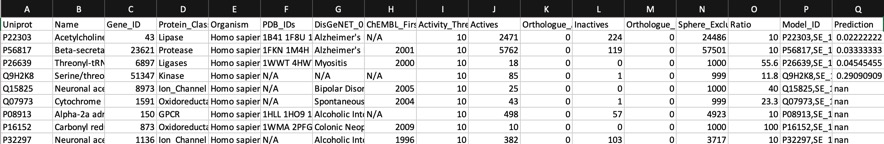
Using AD=0
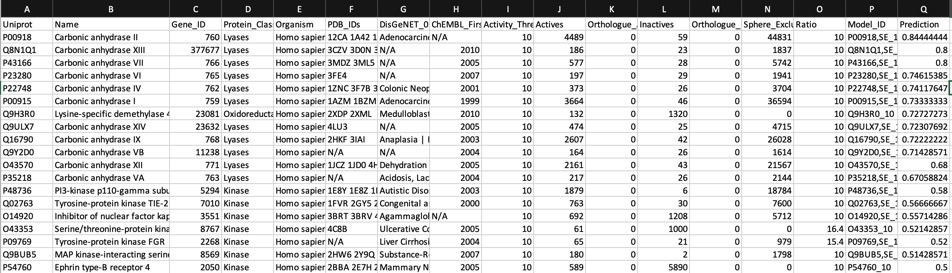
Looking at all the models it is clear that a variety of kinase targets are flagged together with Carbonic anhydrase. A quick literature search confirms that Malaria parasite carbonic anhydrase inhibition is a known mechanism of action DOI, and kinase inhibition DOI has been proposed as a therapeutic approach for the treatment of Malaria.
Last Updated 22 January 2019