FieldView is a molecular viewer/editor from Cresset that is designed to show molecules with their associated Field patterns and physicochemical properties. Many molecular visualization/modelling tools seem to assume the charge associated with an atom sits as a point at the center of the nucleus, whilst this makes the computation easy it does not really reflect what the electrostatic surface really “looks like”. Cresset have pioneered the use of field point descriptors to give a more accurate description of the charge around an atom and to enable better comparisons and visualization. This has been shown to be particularly important when trying to understand some molecular interactions such as Aryl-Aryl interactions or creating bioisosteric replacements. FieldView provides a richer more informative view of how molecules are likely to behave in biological systems. FieldView enables you to load your structures from SDF and MOL2 files as well as showing results from all Cresset applications. You can import and compare up to 10,000 compounds at once or copy and paste them into FieldView from your favourite drawing package.
In this review I thought I’d look at some ester bioisosteres that have been reported in the literature. I built the ester, ketone and amide shown below in MOE, minimised the structures, calculated the total polar surface area and then exported as an sdf file containing all three structures. FieldView will also read 2D structure files convert them to 3D and minimise the structures.
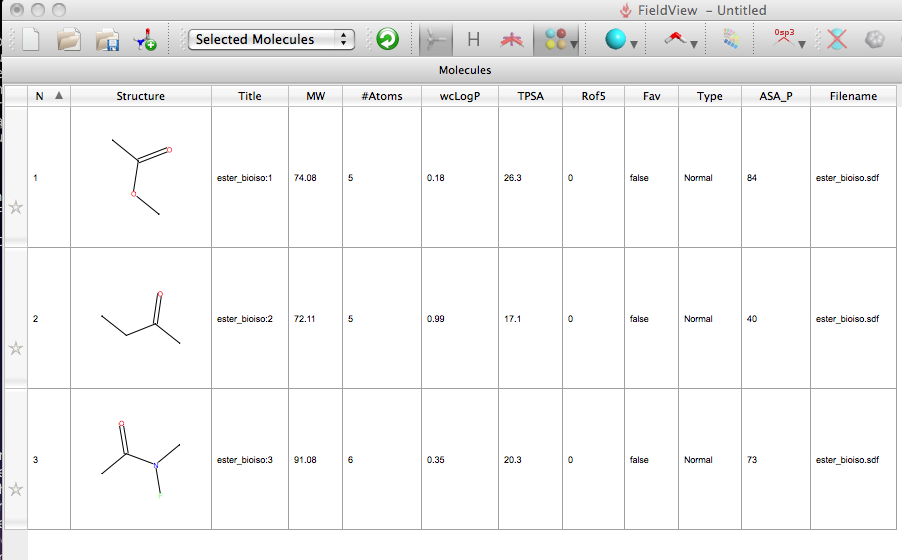
I then opened FieldView and used the “add molecule” command to add the sdf file, all three structures were imported and the molecules displayed in a table, a number of properties were automatically calculated including LogP, MW, number of Lipinski ROF violations, also the total polar surface area calculated in MOE was imported from the sdf file (this is an option in the preferences). You can sort on each of the columns by simply clicking the column header.
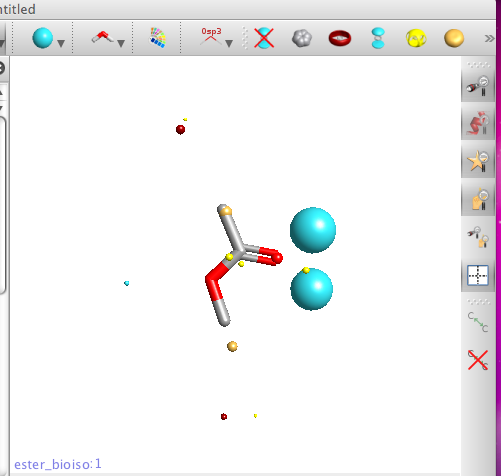
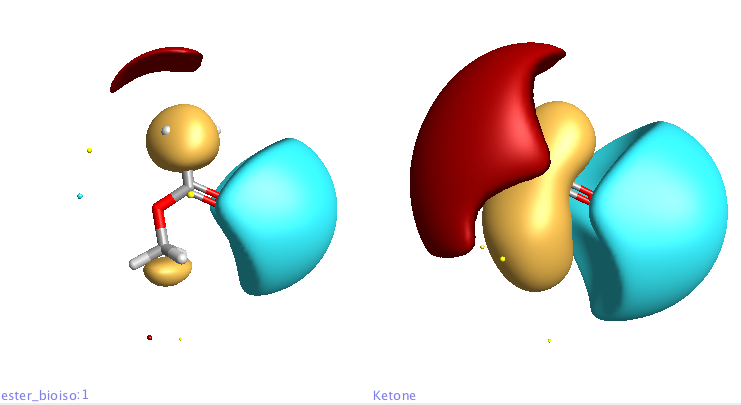
If you then click on one of the structures the molecule the 3D structure is displayed in the structure pane together with the calculated field patterns. Field patterns rely on an accurate and detailed description of atoms in molecules. Traditional technology uses simplified descriptions of atoms and molecules, in particular atom based charges. Cresset’s method relies on XED models of atoms and molecules, which present a more complex, accurate description of the charge around any atom. Note the two blue negative points next to the carbonyl rather than a single negative point inline with carbon oxygen double bond that many applications report. There are also options for bond display style and toggling hydrogen display on or off.
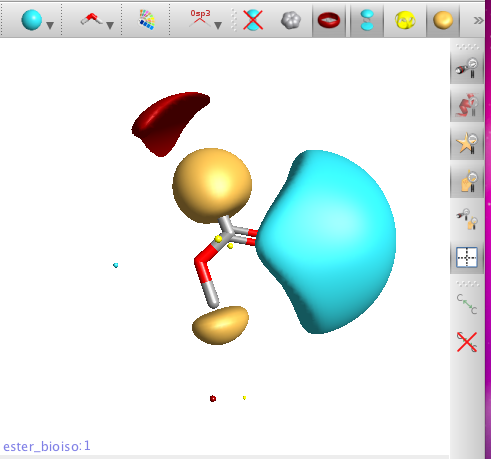
Clicking on the buttons at the top of the molecule view allows the display of hydrophobic surfaces (yellow), and both positive (red) and negative fields (blue), clicking on the button again turns off the appropriate surface.
By selecting multiple structures you can view the corresponding molecules and fields either overlayed or as a grid. In the display below the ester (left) is compared with the ketone (right).
To now add another structure you can simply right-click on an existing structure and choose “edit a copy of the selected molecule” this opens a structure editor that allows modification of the existing structure. When you have completed editing the structure click the minimise button and the structure will be minimised to the nearest local energy minimum and the field patterns added.
The minimiser is XEDMIN, this is based on their proprietary forcefield (FF2), which uses eXtended Electron Distributions (XEDs) to model the electrostatic fields in a more accurate manner. The use of XED based forcefields has been particular successful in studying aromatic interactions (Chessari, G., Hunter, C. A., Low, C. M. R., Packer, M. J., Vinter, J. G. and Zonta, C. (2002), An Evaluation of Force-Field Treatments of Aromatic Interactions. Chemistry – A European Journal, 8: 2860–2867. doi: 10.1002/1521-3765(20020703)8:13<2860::AID-CHEM2860>3.0.CO;2-N) Comparison of the experimental aromatic interaction energies and the X-ray crystal structures of complexes with the calculated data show that conventional molecular mechanics force fields (MM2, MM3, AMBER and OPLS) do not perform well. However, the XED force field which explicitly represents electron anisotropy as an expansion of point charges around each atom reproduces the trends in interaction energy and the three-dimensional structures exceedingly well.
I added the oxime ether shown below. Note the main negative potential is associated with the nitrile not the oxime. It would be nice to be able to rotate the structures independently in order to align the surfaces, at the moment it is probably better to build structures by modification of an existing structure to keep them more or less aligned.
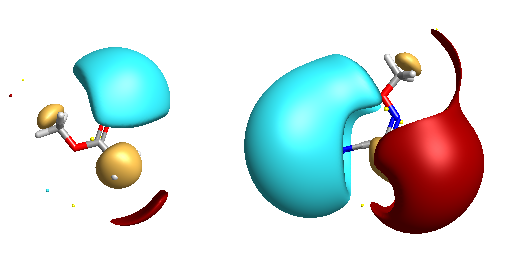
Five-membered heterocycles have been used as bioisosteric replacements for esters on a number of occasions and in particular there are many examples of the 1,2,4-oxadiazole being used. The corresponding field views are shown below, note the negative potentials are associated with the nitrogens of the heterocycle (right).
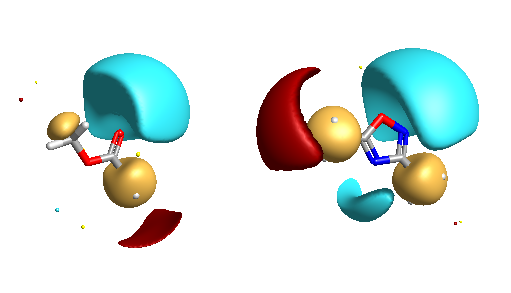
It should be noted that the fields are highly dependent on the conformation of the molecule, with different conformations giving different field shapes. Cresset use another product FieldAlign to do this. This is used for mapping a molecule or a series of molecules to a fixed conformation of the target structure. The generation of multiple conformers is automatically handled by FieldAlign, using a combination of torsion driving and high-temperature dynamics, followed by a short minimisation step to the nearest local minimum. You could of course use an external program to generate multiple conformations of a given structure and then export in sdf format and import into FieldView.
Cresset have also made a number of webclips available that illustrate FieldView features.
Create and examine a molecule series Compare small hetero-aromatic moleculesDrawing new molecules with FieldView
They also provide a couple of case studies.
FieldView is a pretty intuitive application to use and offers access to a unique way of viewing molecules and in particular possible intramolecular interactions, and as such is a valuable addition to the chemists desktop.

One thought on “Review of FieldView”
Comments are closed.