SeeSAR is a new product from BioSolve-it, it is intended as an interactive tool for designing/improving ligands for drug discovery.
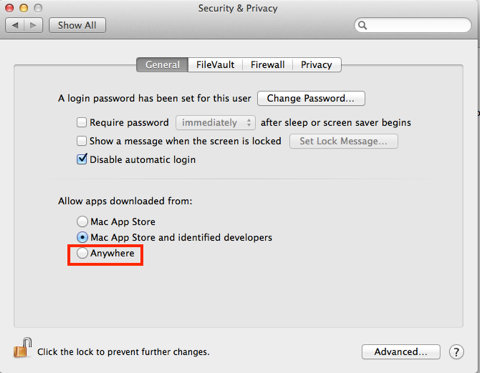
The download and installation is painless but will depend on the security settings, since this is not downloaded from the Mac App Store you will have to temporarily change the Security settings to allow installation. Remember to change them back afterwards.
Starting Up
When you first start up SeeSAR you get an option to download a file from RCSB Protein Data Bank or to open a file. If you are opening an internally generated PDB then there should be no concerns, however I would urge caution if using a file downloaded from the PDB. It may be useful to first check the PDB file using another software package to correct missing or incorrectly assigned residues, then save as a PDB and opened the local file.
Interacting with the protein/ligand
Once opened the structure is displayed in the main screen, the view is clear and rotating the structure is smooth and responsive. The display is fairly minimalist, with two buttons at the top left for structure import, a tab on the left edge containing a ligand list and tab at the bottom right giving access to utilities.
The main controls support the conventional Mac input.
right-click rotate Note: if you click on an object, this is taken as the center of rotation. Otherwise, the rotation is – depending on the viewing mode – either around the center of the entire protein or around the center of the binding site. So don’t just click anywhere but close to a ligand atom of interest to rotate around it.
left-click select
[command] + right-click translate middle-click
[shift] + right-click z-rotation
[command] + [shift] + right-click zoom mouse-wheel
[del] delete table entries [up] and [down] arrows move to the respective next table entry
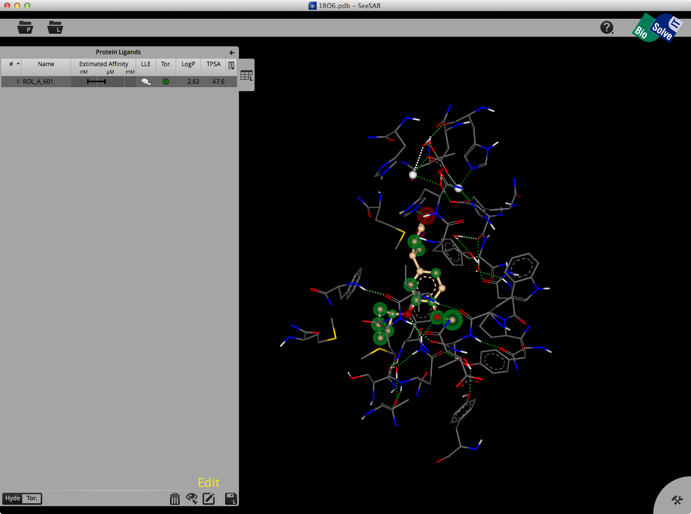
Selecting a ligand from the left hand tab brings up a binding site view of the structure as shown below. The ligand list tab can be modified to include a variety of calculated physicochemical properties, it also gives an estimated affinity and LLE. The structure display is pretty intuitive, green is a favourable interaction, red unfavourable, with the size of the coloured spheres indicating the magnitude of the interaction. Currently there is no option for a protein surface but I understand it is high on the list of things to do.
Clicking on the Edit button brings up another palette attached to the right hand edge, this contains tools to edit the structure. These allow you to change atom or bond types, add atoms, clean modified structures etc. Whilst there is no protein surface display there is a “white fog” surrounding the molecules (see image below), this shows you free, explorable space between ligand and protein. Fog is where at least a fluorine or methyl group would fit between ligand and protein. It is intended as a guide line for the user to improve the tightness of fit.
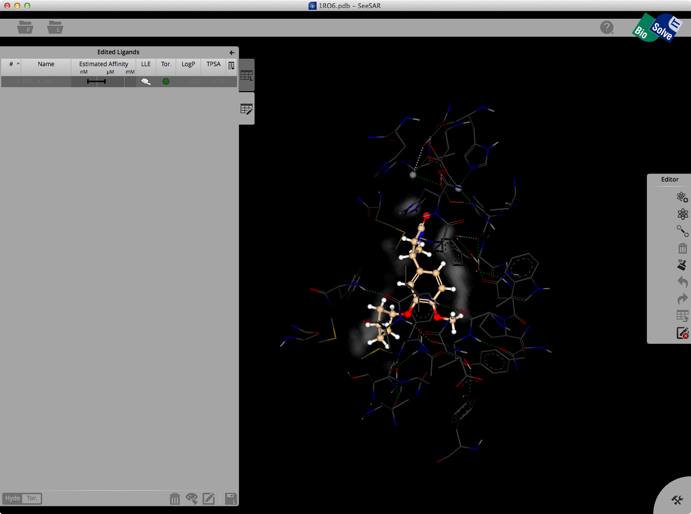
Once you have modified the structure you can save the new ligand to the ligand list, and the structure will be optimised in the active site, the affinity estimated and the atoms colour coded as shown below.
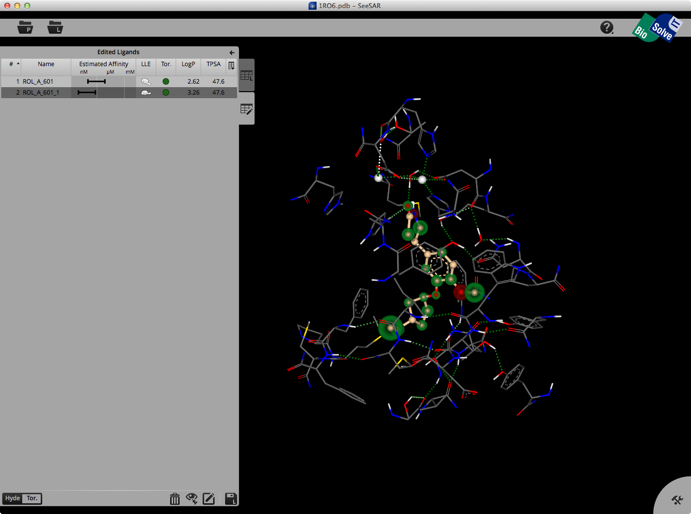
The whole process is quick and easy and the user can rapidly explore new ideas. Unfortunately there is no 2D display of ligands in the ligand list so sometimes it can be difficult to keep track of modifications. You can inspect the contributions of individual atoms by selecting “Atom Labels” from the utilities tab in the bottom right corner and then clicking on an atom.
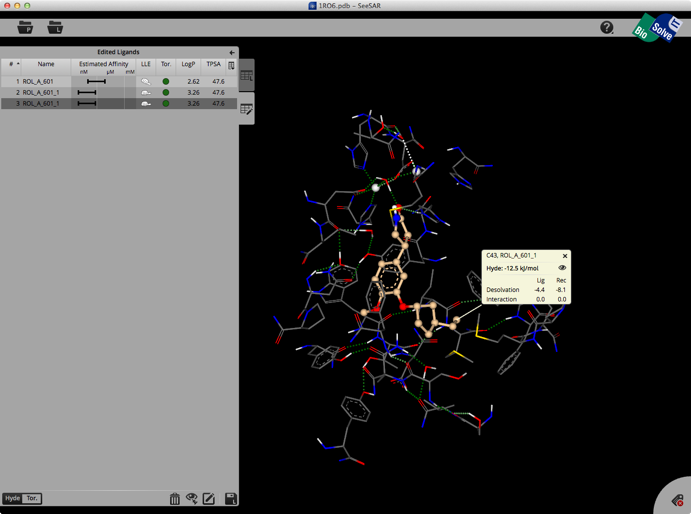
At the bottom of the ligands table you may switch between Hyde-coloring and Torsion-coloring. Red torsions indicate outliers from the CSD-based torsional preferences. At least one red bond also results in the red Tor.-flag in the table.
Summary
This is a really interesting application, it seems a little rough around the edges but the developers have released it early are very keen to get feedback. I found them very responsive and enthusiastic about getting the views of users involved in how the application evolves. I would certainly encourage people to download it and use the free trial period to give it a go.
A couple of points that might be worth being aware of, I downloaded the structure 3K83 from the PDB
http://www.rcsb.org/pdb/explore/explore.do?structureId=3K83
This contains ligand and dodecanol, SeeSAR identifies dodecanol as the ligand and not the “real” ligand, it turns out this is because the ligand form a stabled tetrahedral intermediate with the protein and is thus covalently attached. Unfortunately you cannot edit the structure from within SeeSAR, this also means you cannot remove waters that you might want a modified ligand to displace. As I mentioned above if you are using a structure from the PDB it is worth doing some protein preparation before importing into SeeSAR.
I could not find a way to alter the charges on ligands, so to introduce a protonated amine I had to first create the quaternary nitrogen by adding a methyl, I then deleted the carbon and it was replaced by a hydrogen resulting in a protonated nitrogen. Similarly carboxylates are created by adding the acid then deleting the hydrogen.
Last updated 18 June 2014
One thought on “A Review of SeeSAR”
Comments are closed.