Whilst much computational work is undertaken to support, library design, virtual screening, hit selection and affinity optimisation the reality is that the most challenging issues to resolve in drug discovery often revolve around absorption, distribution, metabolism and excretion (ADME). Whilst we can measure the levels of parent drug in various medium tracking metabolic fate can often be a considerably more difficult proposition requiring significant resources. For this reason prediction of sites of metabolism has become the subject of current interest.
The challenge of the problem should not be under-estimated, there are multiple different potential enzymic reaction types, those that act directly on the drug (phase I) and those the further functionalize metabolites (phase II), any drug can be a substrate for multiple enzymes. Predictions involving QM calculations or docking to the flexible binding sites of cytochrome P450 enzymes require prohibitive computing resources.
FAME DOI is a collection of random forest models trained on a comprehensive and highly diverse data set of 20,000 small molecules annotated with their experimentally determined sites of metabolism taken from multiple species (rat, dog and human). In addition dedicated models are available to predict sites of metabolism of phase I and II processes. Remarkably this is achieved using only 7 easily calculated descriptors (Table 1), six interpretable atomic descriptors (encoding the element type, hybridization state, and electronic configuration of each atom) and one molecular descriptor (encoding the topological size of a molecule).
| Table 1 | |
|---|---|
| Descriptor | Definition |
| PartialTChargeMMFF94 | Total partial charges of a heavy atom as derived from the MMFF94 model |
| PartialSigmaCharge | Gasteiger−Marsili sigma partial charges in sigma-bonded systems |
| PiElectronegativity | Pi Electronegativity |
| SigmaElectronegativity | Gasteiger−Marsili sigma electronegativity |
| SybylAtomType | Sybyl atom type for a specific atom, encoding element type and hybridization state |
| EffectiveAtom Polarizability | Effective atom polarizability of a heavy atom |
| MaxTopDist | Maximum topological distance between two atoms of a molecule |
FAME requires Java 7 as the default JVM, if you get errors it might be worth checking the java version instructions are here.
If now in the Terminal navigate you to “fame-0.9.11-SNAPSHOT/bin” and type
./fame
The following usage message should be displayed:-
FAst MEtabolizer (FAME): A Rapid and Accurate Predictor of Sites of Metabolism
The FAME Development Team
Johannes Kirchmair, jk528@cam.ac.uk
Mark J Williamson, mw529@cam.ac.uk
Andrew Howlett, aph36@cam.ac.uk
Robert C. Glen, rcg28@cam.ac.uk
Git version: 7c549c1
This copy is licensed for academic use only.
Please note that FAME comes with no warranty and redistribution is not permitted.
The license for this software will expire 01-June-2014
**NOTE: INPUT (MOLECULES) MUST HAVE STRONG ACIDS DEPROTONATED AND BASES PROTONATED.
HYDROGENS MUST BE PRESENT. THIS CAN BE DONE USING THE -p OPTION IN OPEN BABEL (FREE SOFTWARE)
HYDROGENS CAN ALSO BE ADDED AUTOMATICALLY USING THE -h OPTION WITHIN FAME
BUT THIS HAS NOT BEEN TESTED AND IS NOT RECOMMENDED.**
Usage:
-i <input file> path to input file (sd file)
-m <model> model
all general (global) model, phase 1 and phase 2 metabolism
allPhase1 general (global) model, phase 1 metabolism only
allPhase2 general (global) model, phase 2 metabolism only
human human metabolism, phase 1 and phase 2
humanPhase1 human metabolism, phase 1 only
humanPhase2 human metabolism, phase 2 only
rat rat metabolism, phase 1 and phase 2
ratPhase1 rat metabolism, phase 1 only
ratPhase2 rat metabolism, phase 2 only
dog dog metabolism, phase 1 and phase 2
dogPhase1 dog metabolism, phase 1 only
dogPhase2 dog metabolism, phase 2 only
-h add explicit hydrogens
-v visualise results using Jmol (optional)
-vm visualise results after minimising conformation (optional)
Please note that the visualiser, Jmol, will show the first molecule on startup.
You can select other molecules ("models") from the context menu, which is called by
right click from within the 3D viewer.
Example for Windows
fame.bat -i ..\ExampleData\diazepam.sdf -m human -v
Example for Mac and Linux:
./fame -i ../ExampleData/diazepam.sdf -m human -v
FAME is very fast. Measured for a batch of 200 druglike molecules calculated on a 2011 MacBook Air with an Intel i7 dual-core CPU, descriptor calculation takes about 2.2 s per molecule (110 ms per heavy atom), and the SoM prediction process itself only takes about 80 ms per molecule.
Predicting the metabolism of Diazapem
The download includes the structure of diazepam so I thought I’d use that to start with.
Diazepam and a number of the major metabolites, nordazepam, temazepam and oxazepam, have been measured in human urine samples by liquid chromatography DOI.
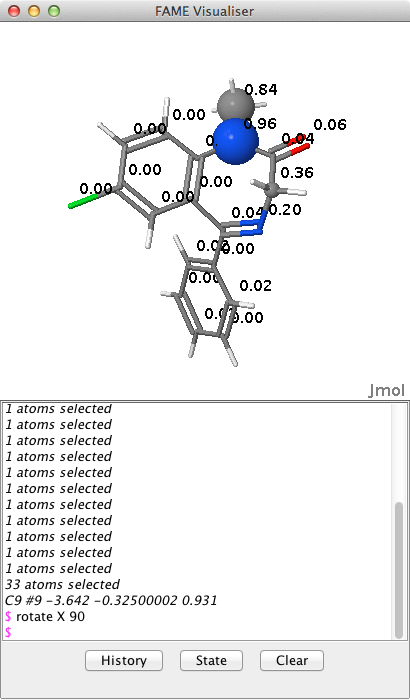
The results of the prediction using FAME are shown below, with the most likely sites for metabolism highlighted with spheres, the radius of the sphere reflecting the probability of metabolism. Clicking on atoms updates the lower panel with the coordinates of atoms. The output from this panel is not relevant for the user but the user can type in commands into this panel if needed e.g.
rotate X 90
More Jmol commands can found here JMOL docs.
However in more congested systems it might be useful to be able to click on an atom and have the atom type, coordinates and the FAME score displayed.
he calculation also produces two files, a .csv file that includes the descriptor values (columns C to I) for all molecules plus the predictions from FAME (last column). There is also a “Class label” which is apparently used for known sites of metabolism. The other file has the extension .arff, this file is only relevant for users who wish to directly import this data into WEKA for further analysis.
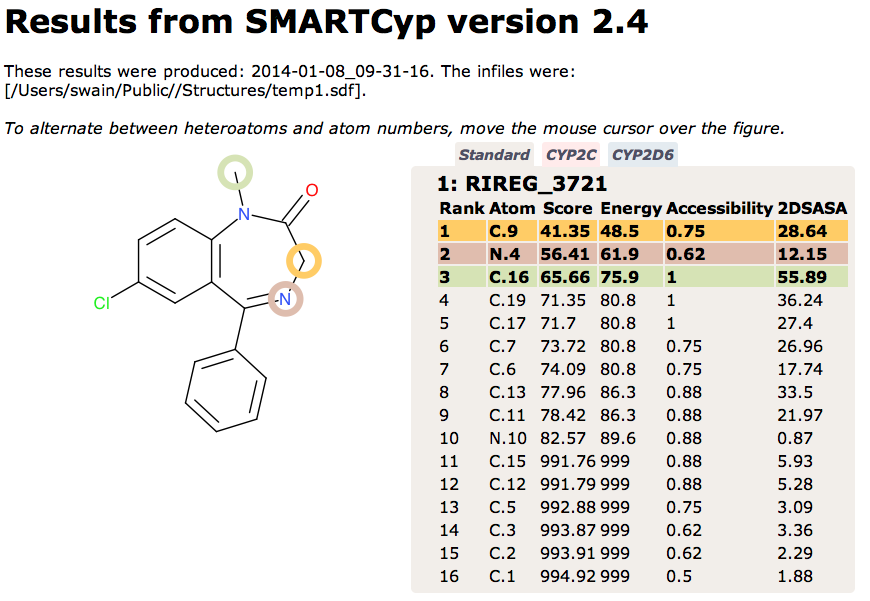
Temazepam formation is carried out mainly by CYP3A isoforms, whereas the formation of N-desmethyldiazepam is mediated by both CYP3A isoforms and S-mephenytoin hydroxylase Br J Clin Pharmacol. 1994 August; 38(2): 131–137 Since these metabolites are derived from CYP mediated metabolism I thought it might be interesting to compare with SMARTCyp a computational tool for specifically predicting for CYP mediated metabolism. SMARTCyp is a reactivity-based model for human cytochrome P450-based metabolism. It computes likely sites of metabolism by estimating transition state energies from a library of precalculated fragments. SMARTCyp also uses span descriptors accounting for steric accessibility and functional group interactions allowing models to be built for specific CYP isoforms..
SMARTCyp P. Rydberg, D. E. Gloriam, J. Zaretzki, C. Breneman and L. Olsen, ACS Med. Chem. Lett., 2010, 1, 96-100 , P. Rydberg, D. E. Gloriam and L. Olsen, Bioinformatics, 2010, 26, 2988-2989, P. Rydberg and L. Olsen, ACS Med. Chem. Lett., 2012, 3, 69-73, P. Rydberg and L. Olsen, ChemMedChem, 2012, 7, 1202-1209, P. Rydberg et al., Angew. Chem, Int. Ed. 2013, 52, 993-997 and P. Rydberg et al., Mol. Pharmaceutics 2013, 10, 1216-1223.
The results are shown below, the results are broadly similar, however SMARTCyp appears to place greater emphasis on the N4 oxidation, formation of the N-oxide is known.
Non-CYP mediated metabolism
Aldehyde oxidase (AOX1) (EC1.2.3.1) is a non-NADPH dependent, molybdenum cofactor containing soluble enzyme present in the liver and other tissues of several mammalian species, and whilst Cytochrome P450 enzymes have been extensively studied and for part of most drug discovery cascades it worth remembering they are not the only sources of xenobiotic metabolism. Aldehyde oxidase might be of particular interest for certain chemotypes, because in addition to the oxidation of aldehydes to carboxylic acids it is also responsible for the oxidation of a number of nitrogen-containing heterocyclic systems.
Recently the development of the Selective MET receptor tyrosine kinase SGX523 was halted due to AOX1 mediated metabolism seen in human studies but also renal toxicity that might be caused by the very poor solubility of the AOX1 derived metabolite DOI.
Regrettably, a phase I clinical trial to evaluate the safety of SGX523 had to be discontinued due to kidney toxicity. This unexpected effect is thought to have resulted from accumulation of a metabolite in humans, which was not observed at significant levels during animal toxicology studies. The suspect metabolite, which does not inhibit MET, is highly insoluble and may have crystallized in renal tissue.
There have been shown to be species differences for AOX1 in addition Aldehyde Oxidase is a cytosolic enzyme and so most liver microsomal assays may under estimate the contribution AOX1 may make towards clearance of a compound.
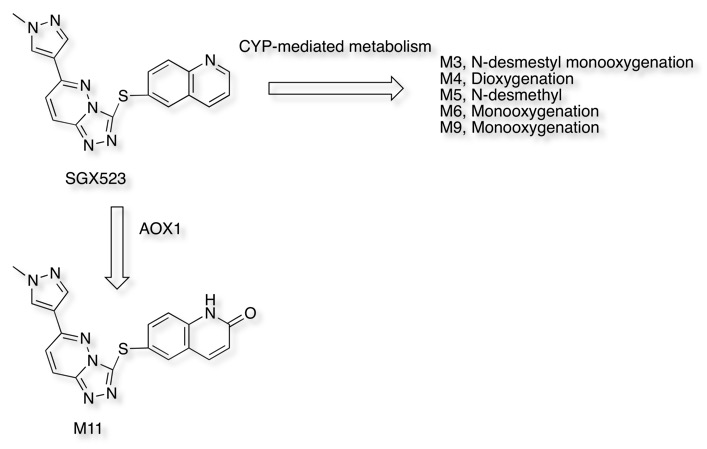
If we first look at the prediction from SMARTCyp it correctly identifies the most likely sites for CYP mediated oxidative metabolism but as expected does not identify the non-CYP mediated metabolism leading to M11
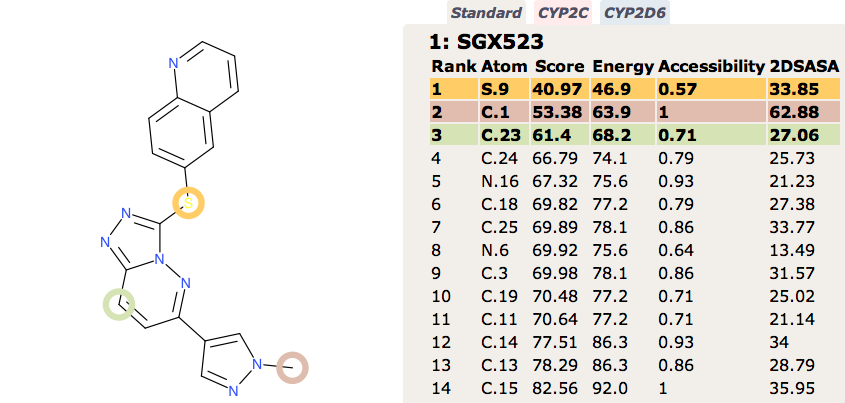
In contrast FAME correctly identifies the potential for oxidation of the quinoline ring to form the poorly soluble quinolone M11. However FAME does not highlight the potential for oxidation of the sulphur.
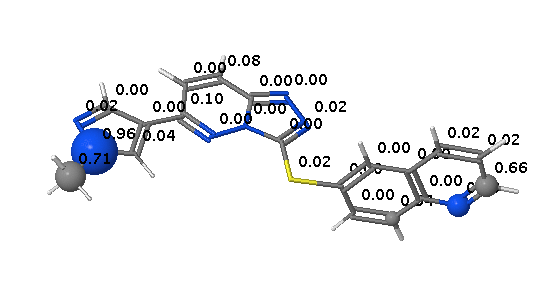
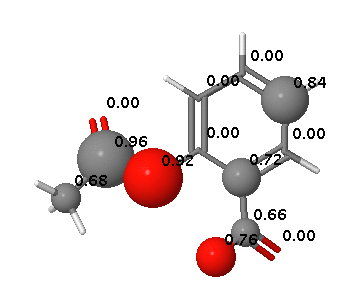
Phase II metabolism
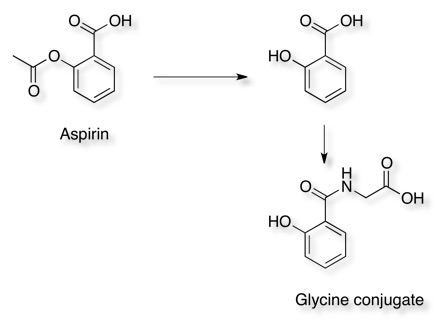
Aspirin hydrolyses to produce salicylic acid (2-hydroxybenzoic acid), Phase II metabolism results in conjugates with glycine (to form salicyluric acid) or glucuronic acid to make several ionised metabolites that can then be excreted in the urine.
Since none of the processes described above are CYP mediated it is not surprising that they are not highlighted by SMARTCyp.
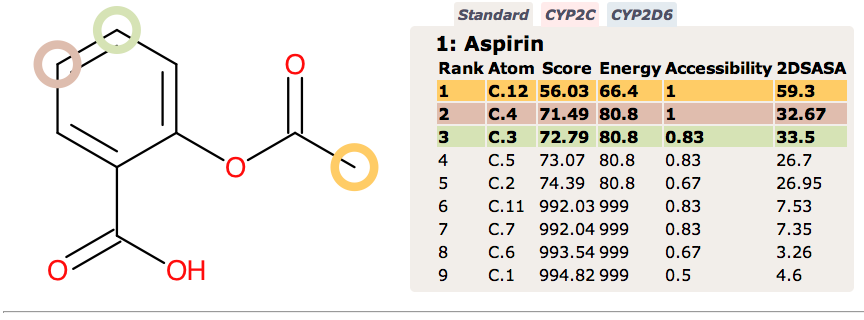
In contrast FAME correctly identifies hydrolysis of the ester and the potential of the carboxylic acid to be involved in metabolic processes.
Summary
FAME offers a high performance prediction of sites of metabolism mediated by a wide variety of mechanisms, and will be an invaluable tool for drug discovery. In terms of prediction of CYP mediated metabolism, I found SMARTCyp scored slightly higher, but FAME was certainly highly competitive. FAME however also offers prediction of non-CYP mediated Phase I metabolism and Phase II metabolism. FAME has certainly set the bar high for subsequent software packages but perhaps more excitingly there are clear opportunities for further development. It would be nice to have the type of reaction annotated, oxidation, hydrolysis, acylation etc. It would also be useful to have an idea of the name of the enzyme/isoform involved. And finally, to have some idea of the likely rate of reaction.
Last Updated 13 January 2014