I’ve just signed up for the meeting, hope to see you there at Burlington House, London, UK Wednesday 8th April 2026 A one-day conference on Chemical
Tag: cheminformatics
Mole2D is a lightweight molecule drawing package written in Python and PyQt6. All code is on GitHub https://github.com/sfmlab/mole2d and it requires RDKit and PyQt6.. PubChem

An update to RDKit has been released 2026_03_1 (Q1 2026). Highlights Note there are a number of backwards incompatible changes. Full details of the release
Sometimes target identification studies can just turn up a list of Uniprot IDs, whilst there are a number of places you can go to find

This meeting will be held at Burlington House, London, UK on Wednesday 8th April 2026. A one-day conference on Chemical Structure Representations will bring together
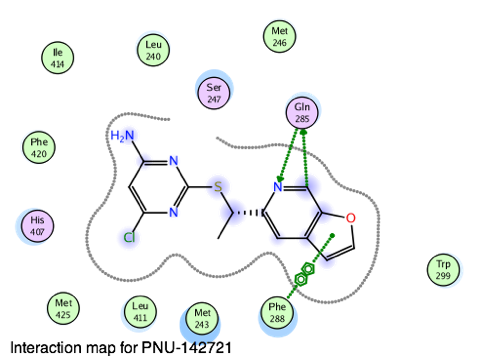
The next OpenADMET blind challenge focuses on predicting human Pregnane-X Receptor (hPXR) induction. The pregnane X receptor (hPXR) is the major determinant of CYP3A gene regulation by
ChemeleonSMD distills the CheMeleon pretrained Directed Message Passing Neural Network (DMPNN) into a SCORE-style architecture running natively on MLX. The SCORE Euler skip connection replaces ChemProp’s hard-overwrite message passing
Recently a guest post from NVIDIA described GPU-Accelerated Clustering with nvMolKit that uses CUDA. A recent post no describes a port of the nvMolKit (CUDA) molecular clustering
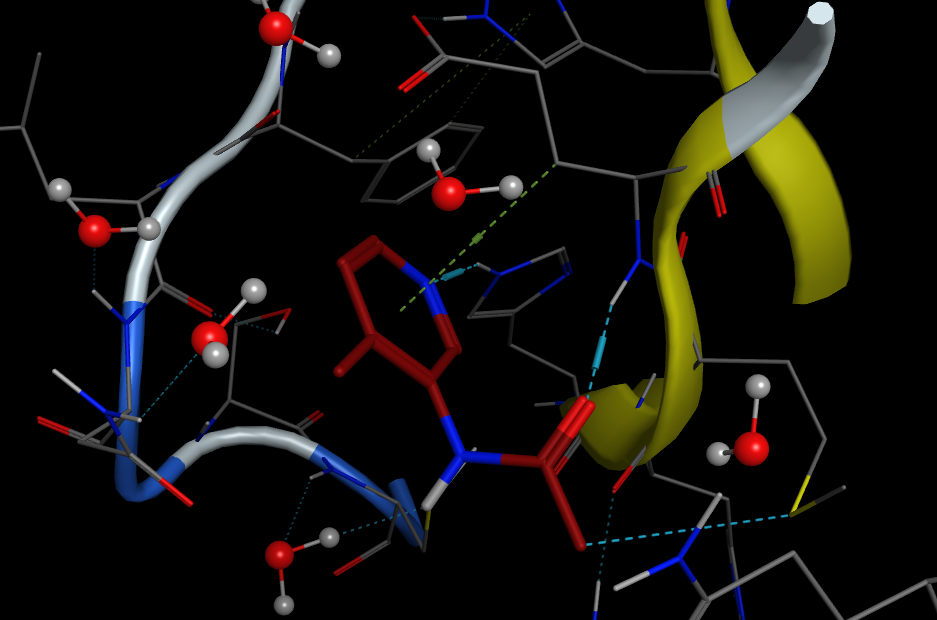
In 2020 as a result of lockdown I was asked to help create a course for MRes students as an introduction to computer-aided drug design.
AI/ML have reignited our thoughts on how we represent chemical structures and so it is very timely that we have a conference on Structural Representation.