In the previous workflow I described docking a set of ligands with known activity into a target protein, in this workflow we will be using a set of ligands from the ZINC dataset searching for novel ligands.
Whilst high-throughput screening (HTS) has been the starting point for many successful drug discovery programs the cost of screening, the lack of accessibility of a large diverse sample collection, or low throughput of the primary assay may preclude HTS as a starting point and identification of a smaller selection of compounds with a higher probability of being a hit may be desired. Directed or Virtual screening is a computational technique used in drug discovery research designed to identify potential hits for evaluation in primary assays. It involves the rapid in silico assessment of large libraries of chemical structures in order to identify those structures that most likely to be active against a drug target. The in silico screen can be based on known ligand similarity or based on docking ligands into the desired binding site.
The clean lead-like set (p.mwt <= 350 and p.mwt >= 250 and p.xlogp <= 3.5 and p.rb <= 7) from ZINC contains around 4.5 million structures. Whilst we could simply dock the entire 4.5 million structures for this exercise we will be using a representative selection. To select these compounds we import all the structures into Vortex and then cluster and subsequently select the centroid of each cluster as described on this pageto yield a set of 50,000 molecules. A set of reasonable conformations for each structure were then generated using the Jupyter notebook as described previously and docked into the target protein using Smina.
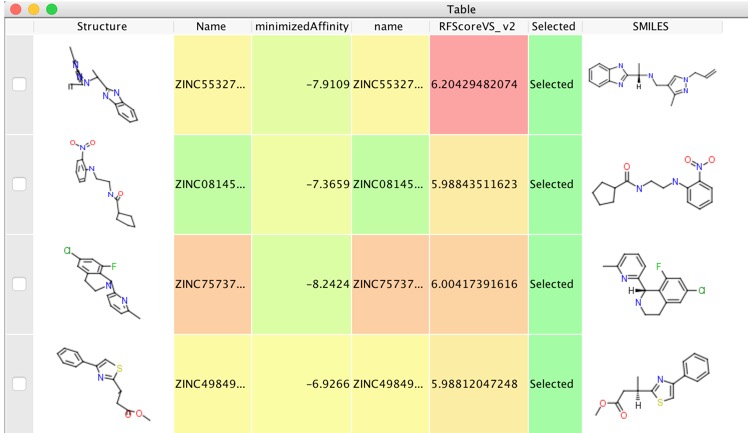
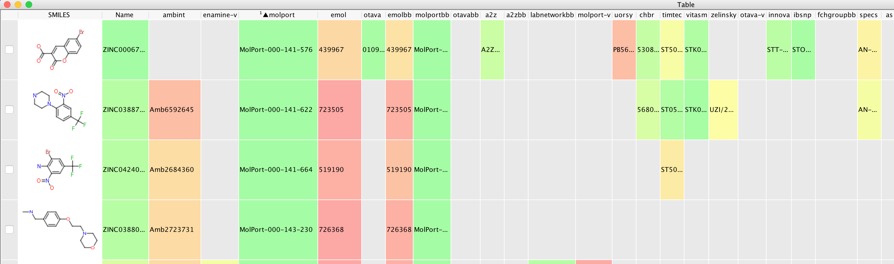
This resulted in 1.9 million docked structures, these were imported into Vortex and the docked structures rescored using RF-Score-VS as described in the previous workflow. We then select the highest scoring poses for each structure and export them to a new workspace.
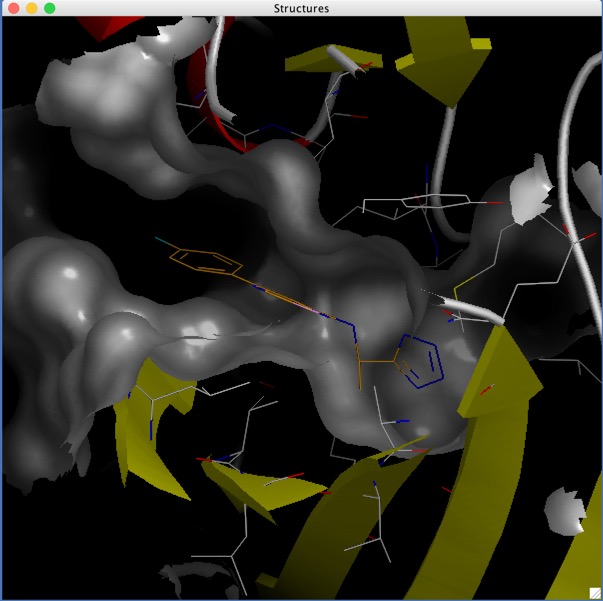
With a workspace containing the selected docking results we can use the embeded AstexViewer to look at the docked poses.
Flagging unwanted structures
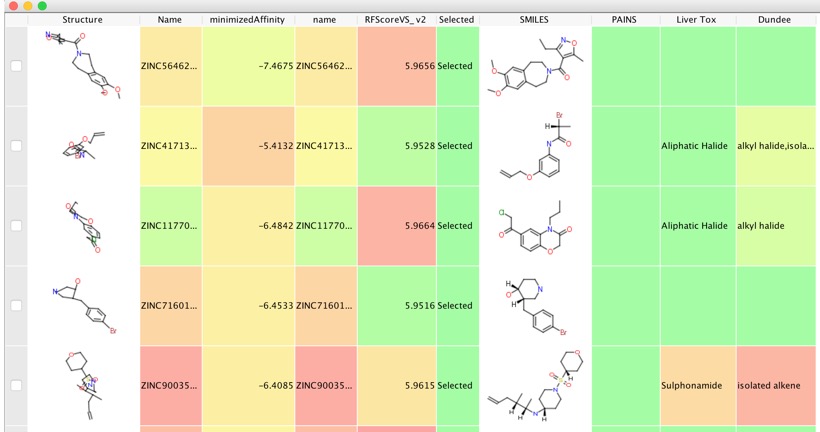
You can now select the some of the top scoring ligands from the virtual screen for purchase and biological evaluation. I took the top 11,000 and exported them to a new workspace, then ran a series of filters to flag potential PAINS, Reactive Molecules, and Hepatotoxic Scripts are here.
These molecules — pan-assay interference compounds, or PAINS — have defined structures, covering several classes of compound. But biologists and inexperienced chemists rarely recognize them. Instead, such compounds are reported as having promising activity against a wide variety of proteins. Time and research money are consequently wasted in attempts to optimize the activity of these compounds. Chemists make multiple analogues of apparent hits hoping to improve the ‘fit’ between protein and compound. Meanwhile, true hits with real potential are neglected….Most of all, academic drug discoverers must be more vigilant. Molecules that show the strongest activity in screening might not be the best starting points for drugs. PAINS hits should almost always be ignored. Even trained medicinal chemists have to be careful until they become experienced in screening. Take it from us: do not even start down these treacherous routes.
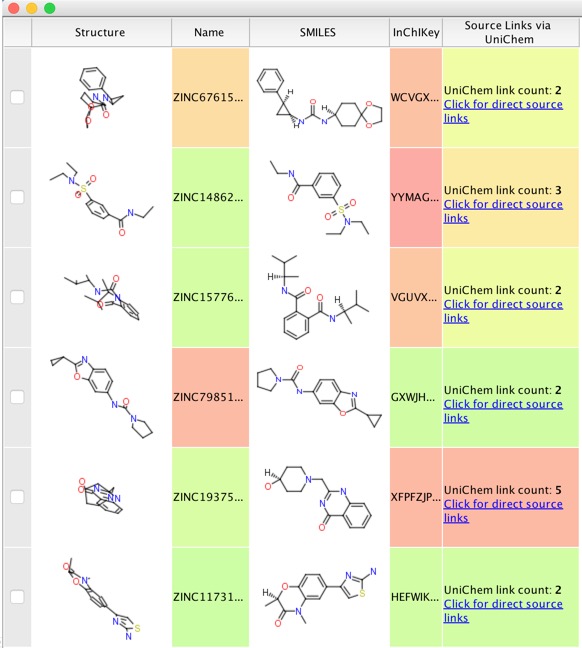
I removed some of the flagged molecules to leave just over 10,000 molecules, we can check if any of these have known biological activity by using the UniChem datasource using the Un1Chem vortex script
Un1Chem is a new web resource provided by the EBI, it is a ‘Unified Chemical Identifier’ system, designed to assist in the rapid cross-referencing of chemical structures, and their identifiers, between database
Whilst the compounds with one or two UniChem links (one presumably the ZINC ID) are fine, if the are 8 or 9 UniChem links it is probably worth clicking on the link to see if the molecule has significant activity at other sites that might be an issue and remove the molecule. Once the final selection has been made I usually export them to a new sdf file and then reopen the sdf file in Vortex. This avoids an issue of accidentally deleting earlier work.
Get the list of vendors for each molecule
Once we have the desired set of docking results we can go about ordering them. Zinc contains molecules from nearly 200 vendors so first we need to find out which vendors have each of the docked ligands. The following Vortex script gets the supplier information for each molecule from ZINC using the ZincID as the search term. The ZINC command language is described in detail here http://wiki.bkslab.org/index.php/ZINC:Command_language.
We can search for the details on a single compound using a search string in this format
The first part of the script asks the user to select the column containing the ZINC ID, then the search string is created and finally the data returned is added to the workspace.
#Get suppliers from ZINC
#All rights reserved.
# Python imports
import urllib2
import urllib
from com.xhaus.jyson import JysonCodec as json
# Standard Vortex imports
import com.dotmatics.vortex.util.Util as Util
import com.dotmatics.vortex.mol2img.jni.genImage as genImage
import com.dotmatics.vortex.mol2img.Mol2Img as mol2Img
import jarray
import binascii
import string
import os
input_label = swing.JLabel("ZINC ID (for input)")
input_cb = workspace.getColumnComboBox()
panel = swing.JPanel()
layout.fill(panel, input_label, 0, 0)
layout.fill(panel, input_cb, 1, 0)
ret = vortex.showInDialog(panel, "Choose ZINC column")
if ret == vortex.OK:
input_idx = input_cb.getSelectedIndex()
if input_idx == 0:
vortex.alert("you must choose a column")
else:
col = vtable.getColumn(input_idx - 1)
rows = vtable.getRealRowCount()
for r in range(0, int(rows)):
zincID = col.getValueAsString(r)
try:
mystr = "http://zinc15.docking.org/substances/" + zincID + "/catitems/subsets/for-sale.json?count=all"
myreturn = urllib2.urlopen(mystr).read()
j = json.loads(myreturn)
for supplier_data in j:
code = supplier_data['supplier_code']
supplier = supplier_data['catalog_short_name']
resultCol = vtable.findColumnWithName(supplier, 1)
resultCol.setValueFromString(r, code)
except Exception:
continue
Selecting Vendors
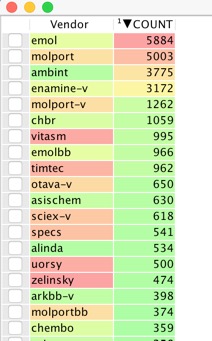
With so many vendors it is a bit daunting trying to decide on which ones to order from, the following script gives you a new worksheet contains a list of all vendors and how many molecules are available from each.
The first part of the script builds a dialog box to allow user to select preferred vendors (or you can just select them all). Then loop through each selected vendor and count how many records there are in the column, then create new workspace with selected vendors and number of molecules available from each.
#Getting a list of suppliers for ordering compounds
# Python imports
import urllib2
import xml.etree.ElementTree as etree
# Vortex imports
import javax
import com.dotmatics.vortex.util.Layout as layout
import com.dotmatics.vortex.components as components
import time
import com.dotmatics.vortex.util.Util as Util
import com.dotmatics.vortex.mol2img.jni.genImage as genImage
import com.dotmatics.vortex.mol2img.Mol2Img as mol2Img
import jarray
import binascii
import string
import os
if vws is None:
vortex.alert("You must have a workspace loaded...")
# Get list of suppliers and get user to select vendors
columnNames = vtable.getColumnNames(0)
cols = []
includeCols = []
for name in columnNames:
cols.append(name)
# Prompt user for list of suppliers
content = javax.swing.JPanel()
duallist = components.VortexDualList()
duallist.setItems(cols, None)
layout.fill(content, duallist, 0, 0)
ret = vortex.showInDialog(content, "Choose the suppliers columns")
if ret == vortex.OK:
for i in duallist.getItems():
includeCols.append(cols[i])
for col in includeCols:
cols.remove(col)
if not len(includeCols):
vortex.alert("No suppliers selected")
sys.exit(0)
Results =[]
for col in includeCols:
#count number of records for each vendor
Vendortotal = 0
mycol = vtable.findColumnWithName(col)
for r in range(vtable.getRealRowCount()):
if (mycol.getValueAsString(r)) != 'N/A':
Vendortotal = Vendortotal + 1
#Need to create array VendorName,count
Results.append([col, Vendortotal])
# Output to new table
#arrayToWorkspace(results, properties, 'My New Workspace')
#Python arrays can be turned into a new workspace using the new utility function, arrayToWorkspace. This takes a 2D array of data, a 1D array of column names and a string for the new workspace name
arrayToWorkspace(Results, ['Vendor', 'COUNT'], 'Vendor Analysis Output')
You may have preferred vendors and depending on your purchasing department you may have vary things but this is one possible workflow.
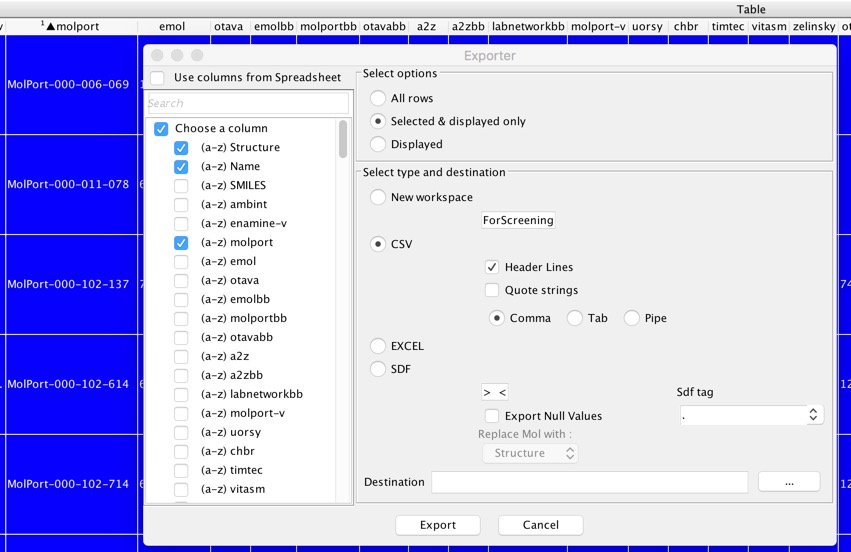
Select one of the major suppliers, sort the workspace containing all the selected docked ligands to bring all the molecules from that vendor to the top of the list, then select all the molecules available from that vendor. Now select “Export” and in the dialog box elect the columns you want to export, check the “Selected & displayed only” radio button and the CSV radio button. You can now export the highlighted records to a file (or another workspace).
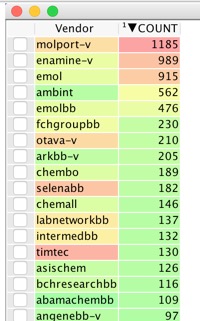
If you now delete the highlighted records (this is why I work on an exported sdf) and run the Vendor Analysis script again, this runs the analysis over the remaining compounds and you can see which vendor can supply the remaining compounds. The process can be repeated until you have exhausted the selected docked ligands.
With the exported files containing the molecules and ID for each vendor you can now order the compounds. People often underestimate how laborious the selection and ordering process can be but I hope this workflow makes it a little less painful.
Another option is to take the top scoring molecules and go back to the clustering and select the rest of the cluster that the selected molecule came from and try docking all the members of the cluster to see if any offer an improved score.
This publication is certainly worth reading “Performance of multiple docking and refinement methods in the pose prediction D3R prospective Grand Challenge 2016” DOI.
The largest single factor governing performance of all methods is availability of data on the chemotypes of interest. This largely reflects the inherent problems in non-native binding pose determination. Docking non-native ligand-receptor complexes is significantly more diffcult than docking a compound from a known chemotype into its native receptor. Our best performing methods attempt to account for this difficulty. Ensemble docking employs all available receptor structures to simulate protein flexibility, but is limited by prior knowledge and cannot access receptor conformations not previously captured. Molecular dynamics based methods have the potential to access novel receptor conformations, but are still largely dependent on initial binding pose, though they do have the potential to slightly improve partially correct poses, particularly in regards to receptor conformation. Considering more than one binding mode hypothesis can increase significantly the chance of success, especially for novel ligands.
We use cookies to ensure that we give you the best experience on our website. If you continue to use this site we will assume that you are happy with it.