Prediction of the metabolism of small molecules is very challenging and so having a variety of different tools is always useful. I’ve previously written Vortex scripts to use SMARTCyp and FAME2 and BioTransformer [DOI] is another useful tool to have in the toolbox.
BioTransformer 3.0 is a software tool that predicts small molecule metabolism in mammals, their gut microbiota, as well as the soil/aquatic microbiota. Moreover, it can also assist scientists in the identification of metabolites, which is based on the metabolism prediction. BioTransformer uses both a knowledge-based approach and a machine learning based approach to predict small molecules metabolism. In particular, BioTransformer consists of five independent modules: EC-based, CYP450, Phase II, Human Gut Microbial and Environmental Microbial.
Whilst it is possible to access BioTransformer via the web server (https://biotransformer.ca) I would not submit confidential structures however. Instead it is useful to be able to run the models locally, whilst you could do this from the command line this Vortex script does make things a little easier.
The source code can be downloaded from here and the download should look like this.
You can view details of of how to run it
|
1 |
/usr/bin/java -jar '/Users/chrisswain/Projects/BioTransformer/BioTransformer3.0_20230525.jar' -h |
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90 91 92 93 94 95 96 |
usage: java -jar biotransformer-4.0.jar [-a] [-b <BioTransformer Option>] [-cm <CYP450 Prediction Mode>] [-f <Formulas>] [-h] [-icsv <csv Input>] [-imol <MOL Input>] [-isdf <Sdf Input>] [-ismi <SMILES Input>] [-k <BioTransformer Task>] [-m <Masses>] [-mt <mass threshold for input substrates>] [-multiThread <MultiThread input>] [-ocsv <Csv Output>] [-osdf <Sdf Output>] [-pm <PhaseII Prediction Mode>] [-q <BioTransformer Sequence Option>] [-s <Number of steps>] [-t <Mass Tolerance>] [-useDB <Use retrieving from database feature>] [-useSub <Use the first 14 characters of the InChIkey when retrieving from database feature>] This is the version 4.0 of BioTransformer. BioTransformer is a software tool that predicts small molecule metabolism in mammals, their gut microbiota, as well as the soil/aquatic microbiota. BioTransformer also assists scientists in metabolite identification, based on the metabolism prediction. -a,--annotate Search PuChem for each product, and store with CID and synonyms, when available. -b,--btType <BioTransformer Option> The type of description: Type of biotransformer - EC-based (ecbased), CYP450 (cyp450), Phase II (phaseII), Human gut microbial (hgut), human super transformer* (superbio, or allHuman), Environmental microbial (envimicro)**. If option -m is enabled, the only valid biotransformer types are allHuman, superbio and env. -cm,--cyp450mode <CYP450 Prediction Mode> Specify the CYP450 predictoin Mode here: 1) CypReact + BioTransformer rules; 2) CyProduct only; 3) Combined: CypReact + BioTransformer rules + CyProducts. Default mode is 1. -f,--formulas <Formulas> Semicolon-separated list of formulas of compounds to identify -h,--help Prints the usage. -icsv,--csvinput <csv Input> The input, which can be an csv file. -imol,--molinput <MOL Input> The input, which can be a Mol file -isdf,--sdfinput <Sdf Input> The input, which can be an SDF file. -ismi,--ismiles <SMILES Input> The input, which can be a SMILES string -k,--task <BioTransformer Task> The task to be permed: pred for prediction, or cid for compound identification -m,--masses <Masses> Semicolon-separated list of masses of compounds to identify -mt,--massThreshold <mass threshold for input substrates> Specify the mass threshold used to validate input substrates Default massThreshold is 1500Da. -multiThread <MultiThread input> Please input the parameters using the specific format to run this multi-thread feature -ocsv,--csvoutput <Csv Output> Select this option to return CSV output(s). You must enter an output filename -osdf,--sdfoutput <Sdf Output> Select this option to return SDF output(s). You must enter an output filename -pm,--phaseIImode <PhaseII Prediction Mode> Specify the PhaseII predictoin Mode here: 1) BioTransformer rules; 2) PhaseII predictor only; 3) Combined: PhaseII predictor + BioTransformer rules. Default mode is 1. -q,--bsequence <BioTransformer Sequence Option> Define an ordered sequence of biotransformer/nr_of_steps to apply. Choose only from the following BioTranformer Types: allHuman, cyp450, ecbased, env, hgut, and phaseII. For instance, the following string representation describes a sequence of 2 steps of CYP450 metabolism, followed by 1 step of Human Gut metabolism, 1 step of Phase II, and 1 step of Environmental Microbial Degradation: 'cyp450:2; hgut:1; phaseII:1; env:1' -s,--nsteps <Number of steps> The number of steps for the prediction. This option can be set by the user for the EC-based, CYP450, Phase II, and Environmental microbial biotransformers. The default value is 1. -t,--mTolerance <Mass Tolerance> Mass tolerance for metabolite identification (default is 0.01). -useDB <Use retrieving from database feature> Please specify if you want to enable the retrieving from database feature. -useSub <Use the first 14 characters of the InChIkey when retrieving from database feature> Please specify if you want to enable the using first 14 characters of InChIKey when retrieving from database feature. (* ) While the 'superbio' option runs a set number of transformation steps in a pre-defined order (e.g. deconjugation first, then Oxidation/reduction, etc.), the 'allHuman' option predicts all possible metabolites from any applicable reaction(Oxidation, reduction, (de-)conjugation) at each step.(** ) For the environmental microbial biodegradation, all reactions (aerobic and anaerobic) are reported, and not only the aerobic biotransformations (as per default in the EAWAG BBD/PPS system). |
The format for accessing the prediction tool is
|
1 |
java -jar /Users/chrisswain/Projects/BioTransformer/BioTransformer3.0_20230525.jar -k pred -b allHuman -ismi "O[C@@H]1CC2=C(O)C=C(O)C=C2O[C@@H]1C1=CC=C(O)C(O)=C1" -osdf outputfile -s 2 -m "292.0946;304.0946" -t 0.01 -a |
Vortex Script
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90 91 92 93 94 95 96 97 98 99 100 101 102 103 104 105 106 107 108 109 110 111 112 113 114 115 116 117 118 119 120 121 122 123 124 125 |
# BioTransformer # BioTransformer 3.0 A Web Server for Accurately Predicting Metabolic Transformation Products [Submitted in Nucleic Acids Research, Webserver Issue.Apr.2022] # Wishart DS, Tian S, Allen D, Oler E, Peters H, Lui VW, Gautam V, Djoumbou Feunang Y, Greiner R, Metz TO; # doi: https://doi.org/10.1093/nar/gkac313 import sys import com.dotmatics.vortex.util.Util as Util import subprocess import csv import tempfile smiles = '' col = vtable.findColumnWithName('Structure', 0) if (col == None): col = vtable.findColumnWithName('SMILES', 0) if (col == None): vortex.alert('Load a workspace with a Structure or SMILES column please.') quit() else: inputsmiles = col.getValueAsString(cell_row) else: inputsmiles = vortex.getMolProperty(vtable.getMolFileManager().getMolFileAtRow(cell_row), 'SMILES') inputsmiles = inputsmiles.encode('utf-8') #for testing #vortex.alert('SMILES = '+inputsmiles) # Get the list of column names for the table columnNames = vtable.getColumnNames() # Create a set of tabs for the different parts of the user interface tabs = javax.swing.JTabbedPane() # Create the main part of the user interface content = javax.swing.JPanel() layout.fill(content, javax.swing.JLabel("ID Column"), 0, 0) columnID = javax.swing.JComboBox(columnNames) layout.fill(content, columnID, 1, 0) #Arguments input Transformation = ["Phase 1 (CYP450)", "EC-based", "Phase II", "Human Gut", "Env Microbial", "AllHuman", "SuperBio", "MultiBo", "AbioticBio"] CYP450mode = ["Combined", "RuleBased", "CyProduct"] Iterations = ["1", "2", "3"] #transformation input transformations = javax.swing.JComboBox(Transformation) transformations.setSelectedIndex(5) layout.fill(content, transformations, 1, 5) layout.fill(content, javax.swing.JLabel("Transformation"), 0, 5) #CYP450 mode CYP450 = javax.swing.JComboBox(CYP450mode) CYP450.setSelectedIndex(0) layout.fill(content, CYP450, 1, 10) layout.fill(content, javax.swing.JLabel("CYP450 mode"), 0, 10) #Iterations num_iterations = javax.swing.JComboBox(Iterations) num_iterations.setSelectedIndex(0) layout.fill(content, num_iterations, 1, 20) layout.fill(content, javax.swing.JLabel("Number Iterations"), 0, 20) # Add the column content to the tab panel tabs.addTab("Arguments", content) # show the dialog and process the input if the user presses ok ret = vortex.showInDialog(tabs, "Calculate BioTransformations") if ret == vortex.OK: input_idx = columnID.getSelectedIndex() colID = vtable.getColumn(input_idx) taskID = colID.getValueAsString(cell_row) #vortex.alert(taskID) biotrans_index = transformations.getSelectedIndex() biotrans = Transformation[biotrans_index] #vortex.alert(biotrans) numSteps_index = num_iterations.getSelectedIndex() numSteps = Iterations[numSteps_index] #vortex.alert(numSteps) tmpdir = tempfile.mkdtemp() outputfile = tmpdir + "/" + taskID + '.sdf' #for testing #outputfile = '/Users/chrisswain/Public/'+ taskID + '.sdf' #Demo command #java -jar /Users/chrisswain/Projects/BioTransformer/BioTransformer3.0_20230525.jar -k pred -b allHuman -ismi "O[C@@H]1CC2=C(O)C=C(O)C=C2O[C@@H]1C1=CC=C(O)C(O)=C1" -osdf outputfile -s 2 -m "292.0946;304.0946" -t 0.01 -a # You can edit these arguments task = "pred" #biotrans = "allHuman" #numSteps = "2" #Need path to jar file subprocess.call(['/usr/bin/java', '-jar', '/Users/chrisswain/Projects/BioTransformer/BioTransformer3.0_20230525.jar', '-k', task, '-b', biotrans, '-ismi', inputsmiles, '-osdf', outputfile, '-s', numSteps], cwd='Users/chrisswain/Projects/BioTransformer') import com.dotmatics.vortex.table.VortexTableModel as vtm #vortex.alert(outputfile) vortex.loadAnyFile(outputfile) vtable.fireTableStructureChanged() |
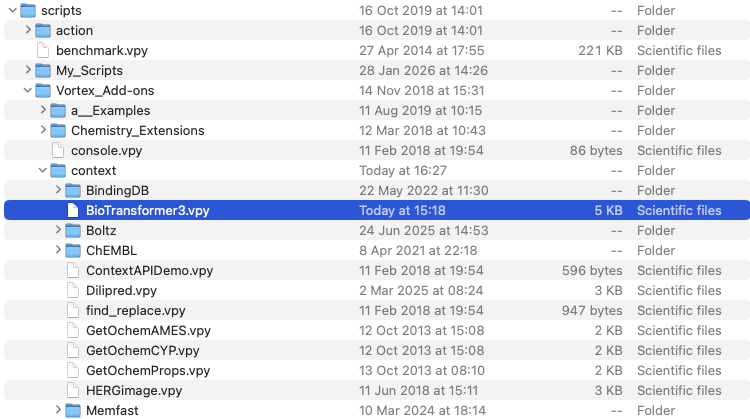
The Vortex script needs to placed in the /Users/username/vortex/scripts/Vortex_Add-ons/context folder as shown below
If you now right-click on the structure in a workspace you should see something like this menu structure.
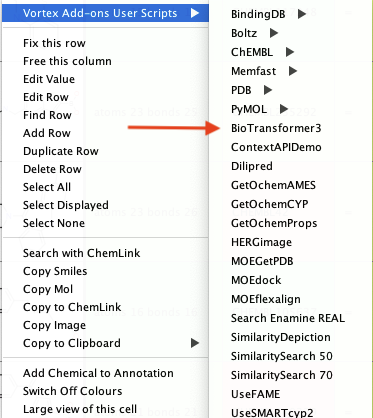
If you now click on BioTransformer3 a dialog box will open
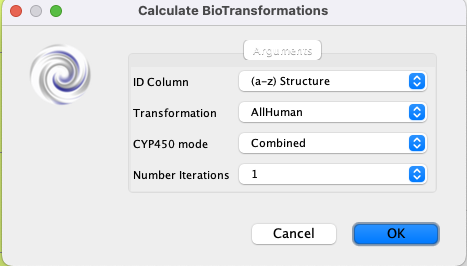
For the dropdown menu “ID Column” choose the field that contains the molecule identifier.
From the “Transformation” dropdown the default is “AllHuman”, but there are options to focus on “Phase I”, “Phase II” or microbial etc.
The “CYP450” allows the user to choose the CYP rule-based prediction mode.
“Number of iterations” is the number of steps to include, Phase 1 only, or to also include phase 2 and subsequent reactions.
There are a number of other options but I decided to try and keep it as simple as possible but still useful. If you click “OK” it will take a few mins to run depending on the complexity of the output.
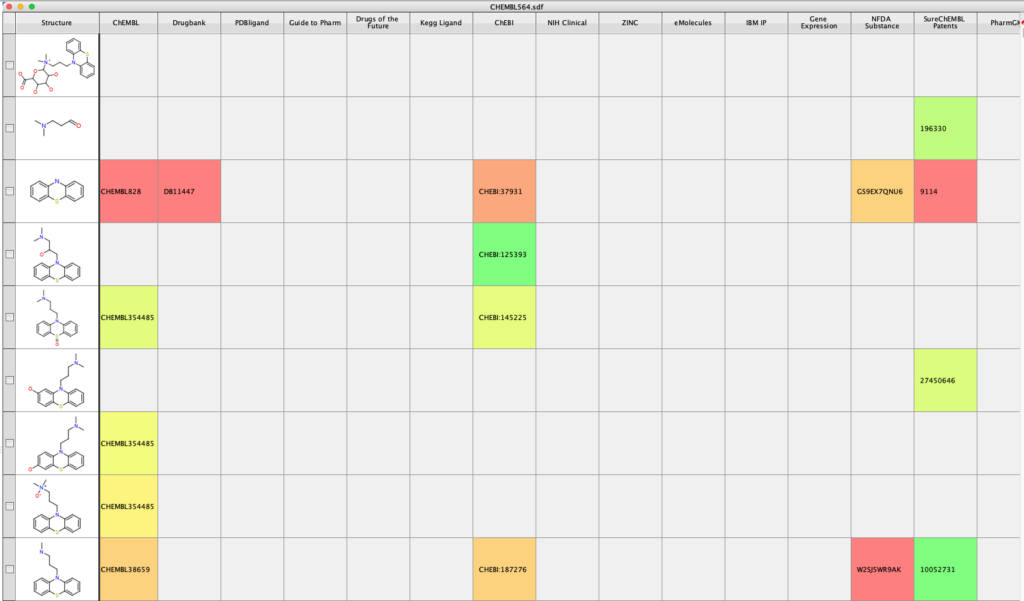
Using this example.
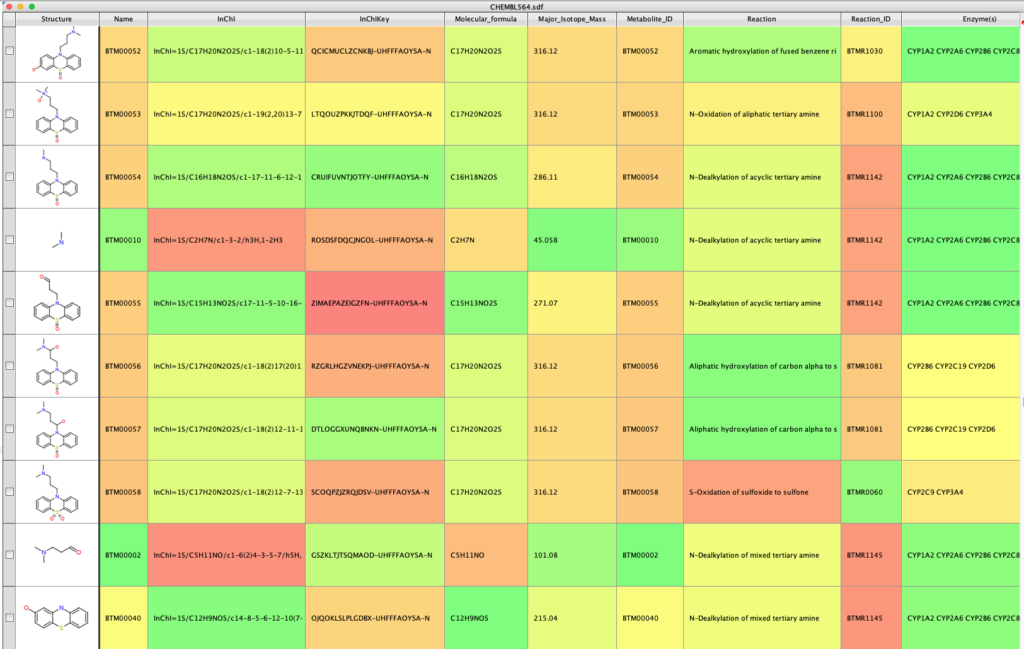
The output look something like this (I’ve hidden some of the empty columns), it contains molecular identifiers (InChi, InChiKey), calculated properties (MWt. Mol Formula) and notes about the metabolic reaction and enzymes that might be involved.
The script can be downloaded from here
With the InChIKey for potential metabolites defined it is also possible look up further information on the molecules, for example you can search using Un1Chem (this is an external search). https://macinchem.org/2023/03/11/vortex-script-for-flexible-search-using-un1chem/.
This will add information from a number of additional datasorces.