I’ve written several posts on the various options for clustering molecules https://macinchem.org/?s=clustering and a recent post from NVIDIA described GPU-Accelerated Clustering with nvMolKit that uses CUDA. This looks very interesting but lies on NVIDIA chips.
A recent post now describes a port of the nvMolKit (CUDA) molecular clustering pipeline to Apple Metal via MLX. All code is on GitHub https://github.com/guillaume-osmo/mlxmolkit?tab=readme-ov-file so I thought I’d have a look.
I first cloned the GitHub repo and then created a conda virtual environment, then cd into the repo and with the conda environment activated installed using pip. The comparison was run using the Jupyter notebook below showing the code used which is based on the scripts in the repo.
Port of the nvMolKit (CUDA) molecular clustering pipeline to Apple Metal via MLX.¶
import numpy as np
import pandas as pd
from rdkit.Chem import PandasTools
from rdkit import Chem
from rdkit.Chem import DataStructs
from rdkit.Chem import rdFingerprintGenerator
from rdkit.Chem import AllChem
import mlx.core as mx
from mlxmolkit.fp_uint32 import fp_uint8_to_uint32
from mlxmolkit.fused_tanimoto_nlist import fused_neighbor_list_metal
from mlxmolkit.butina import butina_from_neighbor_list_csr
#Import molecules
inputsdf = 'Random50Kmols.sdf'
ms = [x for x in Chem.SDMolSupplier(inputsdf,removeHs=False)]
len(ms)
[12:41:16] Warning: ambiguous stereochemistry - overlapping neighbors - at atom 15 ignored [12:41:16] Warning: ambiguous stereochemistry - overlapping neighbors - at atom 15 ignored [12:41:17] Warning: ambiguous stereochemistry - overlapping neighbors - at atom 15 ignored [12:41:17] Warning: ambiguous stereochemistry - overlapping neighbors - at atom 16 ignored [12:41:17] Warning: ambiguous stereochemistry - overlapping neighbors - at atom 7 ignored [12:41:17] Warning: ambiguous stereochemistry - overlapping neighbors - at atom 8 ignored [12:41:17] Warning: ambiguous stereochemistry - overlapping neighbors - at atom 9 ignored
50000
def rdkit_fps_and_bytes(mols, fp_radius: int, fp_nbits: int, n_cpu_threads: int):
"""Generate fps ONCE: return both RDKit fps and packed uint8 bytes for Metal."""
from rdkit import DataStructs
from rdkit.Chem import rdFingerprintGenerator
generator = rdFingerprintGenerator.GetMorganGenerator(radius=fp_radius, fpSize=fp_nbits)
fps = generator.GetFingerprints(mols, numThreads=n_cpu_threads)
nbytes = (fp_nbits + 7) // 8
out = np.zeros((len(mols), nbytes), dtype=np.uint8)
for i, bv in enumerate(fps):
bits = np.zeros((fp_nbits,), dtype=np.uint8)
DataStructs.ConvertToNumpyArray(bv, bits)
out[i] = np.packbits(bits, bitorder="little")[:nbytes]
return fps, out
import time
fp_radius =2
fp_nbits = 2048
cpu_threads = 10
# Fingerprinting (ONCE, shared)
t0 = time.time()
#fps_rdkit, fp_bytes_np = rdkit_fps_and_bytes(ms, args.fp_radius, args.fp_nbits, args.cpu_threads)
fps_rdkit, fp_bytes_np = rdkit_fps_and_bytes(ms, fp_radius, fp_nbits, cpu_threads)
t_fp = time.time() - t0
print(f"Fingerprinting (shared, GetMorganGenerator): {t_fp:.3f}s")
print()
Fingerprinting (shared, GetMorganGenerator): 1.405s
def run_rdkit_workflow(fps, n_mols: int, distance_threshold: float):
"""Blog workflow: BulkTanimotoSimilarity → ClusterData."""
from rdkit.DataStructs import BulkTanimotoSimilarity
from rdkit.ML.Cluster.Butina import ClusterData
t0 = time.time()
distances = []
for i in range(n_mols):
distances.extend(BulkTanimotoSimilarity(fps[i], fps[:i], returnDistance=True))
t_sim = time.time() - t0
t0 = time.time()
clusters = ClusterData(
np.array(distances), n_mols, distance_threshold,
isDistData=True, distFunc=None, reordering=True,
)
t_clust = time.time() - t0
return {
"similarity": t_sim,
"clustering": t_clust,
"total": t_sim + t_clust,
"n_clusters": len(clusters),
"cluster_sizes": sorted([len(c) for c in clusters], reverse=True),
"AllClusters": clusters,
}
# RDKit
distance_threshold = 0.6
n_mols = len(fps_rdkit)
print("--- RDKit (BulkTanimotoSimilarity + ClusterData) ---")
rdkit = run_rdkit_workflow(fps_rdkit, n_mols, distance_threshold)
print(f" Similarity: {rdkit['similarity']:.3f}s")
print(f" Clustering: {rdkit['clustering']:.3f}s")
print(f" Total: {rdkit['total']:.3f}s -> {rdkit['n_clusters']} clusters " f"(largest: {rdkit['cluster_sizes'][0]})")
print()
print(rdkit['AllClusters'][14])
--- RDKit (BulkTanimotoSimilarity + ClusterData) --- Similarity: 179.870s Clustering: 55.766s Total: 235.636s -> 11267 clusters (largest: 188) (38615, 197, 302, 1054, 1417, 4109, 4992, 5765, 5927, 7287, 7822, 8531, 8853, 8876, 9902, 10184, 10280, 10498, 11420, 11979, 12252, 12750, 12797, 12854, 13319, 13512, 14009, 14537, 15333, 15464, 15750, 18385, 18605, 21113, 21487, 22252, 22336, 22386, 23117, 23645, 26018, 26122, 27008, 27366, 27590, 27739, 27831, 29654, 29917, 30142, 30619, 30750, 32043, 32070, 32331, 32445, 33472, 34305, 34707, 35215, 35441, 35892, 36256, 36330, 37617, 38040, 38426, 38742, 39452, 39960, 41513, 41521, 41922, 42286, 42434, 42799, 42892, 42900, 43157, 43177, 43494, 44339, 44514, 44582, 44711, 44756, 44906, 46527, 47571, 48065, 48881, 49277, 49314, 49665)
#now display structures from one of the clusters
from rdkit.Chem import Draw
from rdkit.Chem.Draw import IPythonConsole
m1 = ms[197]
m2 = ms[302]
m3 = ms[1054]
m4 = ms[38615]
m5 = ms[1417]
m6 = ms[4109]
mols=(m1,m2,m3,m4,m5,m6)
Draw.MolsToGridImage(mols)

def run_mlx_workflow(fp_bytes_np: np.ndarray, similarity_cutoff: float):
"""
Full Metal pipeline (like nvMolKit):
1. Fused Tanimoto+threshold → CSR neighbor list (GPU, no N×N matrix)
2. Butina greedy (CPU on CSR)
"""
import mlx.core as mx
from mlxmolkit.fp_uint32 import fp_uint8_to_uint32
from mlxmolkit.fused_tanimoto_nlist import fused_neighbor_list_metal
from mlxmolkit.butina import butina_from_neighbor_list_csr
n_mols = fp_bytes_np.shape[0]
fp_mx = mx.array(fp_bytes_np)
fp_u32 = fp_uint8_to_uint32(fp_mx)
# Fused: Tanimoto + threshold + CSR neighbor list (all on Metal, no N×N matrix)
t0 = time.time()
offsets, indices = fused_neighbor_list_metal(fp_u32, similarity_cutoff)
t_fused = time.time() - t0
# Butina greedy on CPU (CSR)
t0 = time.time()
result = butina_from_neighbor_list_csr(offsets, indices, n_mols, similarity_cutoff)
t_butina = time.time() - t0
n_edges = int(np.diff(offsets).sum())
mem_saved_mb = (n_mols * n_mols * 4) / 1e6
return {
"fused_tanimoto_nlist": t_fused,
"butina": t_butina,
"total": t_fused + t_butina,
"n_clusters": len(result.clusters),
"cluster_sizes": sorted([len(c) for c in result.clusters], reverse=True),
"n_edges": n_edges,
"mem_saved_mb": mem_saved_mb,
"NewClusters": result.clusters
}
# MLX/Metal
distance_threshold = 0.6
similarity_cutoff = 1.0 - distance_threshold
print("--- MLX/Metal (Fused Tanimoto→CSR + Butina CPU) ---")
mlx_res = run_mlx_workflow(fp_bytes_np, similarity_cutoff)
print(f" Fused sim→CSR: {mlx_res['fused_tanimoto_nlist']:.3f}s (Metal, no N×N matrix)")
print(f" Butina: {mlx_res['butina']:.3f}s (CPU CSR greedy)")
print(f" Total: {mlx_res['total']:.3f}s -> {mlx_res['n_clusters']} clusters " f"(largest: {mlx_res['cluster_sizes'][0]})")
print(f" Edges: {mlx_res['n_edges']:,} | Memory saved: {mlx_res['mem_saved_mb']:.0f} MB (no sim matrix)")
print()
--- MLX/Metal (Fused Tanimoto→CSR + Butina CPU) --- Fused sim→CSR: 5.589s (Metal, no N×N matrix) Butina: 0.253s (CPU CSR greedy) Total: 5.842s -> 11213 clusters (largest: 188) Edges: 809,760 | Memory saved: 10000 MB (no sim matrix)
print(mlx_res['NewClusters'][14])
(41794, 571, 1951, 2074, 2742, 2866, 3349, 3629, 3948, 4922, 5067, 5217, 5481, 5484, 7786, 8208, 8769, 8985, 9596, 10002, 10391, 10403, 10429, 10671, 11352, 11640, 11904, 12996, 15851, 16354, 16494, 17128, 17891, 18131, 19655, 20027, 20093, 20259, 20735, 21874, 21955, 22266, 22437, 22861, 23481, 23657, 24173, 24231, 24248, 24373, 24393, 24479, 24597, 24619, 24730, 24834, 24963, 26777, 27253, 27634, 27755, 27904, 29863, 30225, 35053, 36784, 39335, 39393, 39598, 39703, 39880, 39963, 40545, 40652, 40904, 41646, 41752, 42061, 42114, 43148, 43213, 43338, 43374, 43389, 43458, 43747, 44099, 45924, 46054, 47406, 47541, 48315, 48636, 48777)
import pandas as pd
from rdkit.Chem import PandasTools
dataframe = PandasTools.LoadSDF(inputsdf,molColName='ROMol', includeFingerprints=True)
dataframe.head(5)
[14:44:21] Warning: ambiguous stereochemistry - overlapping neighbors - at atom 15 ignored [14:44:22] Warning: ambiguous stereochemistry - overlapping neighbors - at atom 15 ignored [14:44:22] Warning: ambiguous stereochemistry - overlapping neighbors - at atom 15 ignored [14:44:22] Warning: ambiguous stereochemistry - overlapping neighbors - at atom 16 ignored [14:44:22] Warning: ambiguous stereochemistry - overlapping neighbors - at atom 7 ignored [14:44:22] Warning: ambiguous stereochemistry - overlapping neighbors - at atom 8 ignored [14:44:22] Warning: ambiguous stereochemistry - overlapping neighbors - at atom 9 ignored
| Name | ID | ROMol | |
|---|---|---|---|
| 0 | ZINC50764925 | ZINC50764925 | <rdkit.Chem.rdchem.Mol object at 0xcb08486d0> |
| 1 | ZINC65292537 | ZINC65292537 | <rdkit.Chem.rdchem.Mol object at 0xf4e8694d0> |
| 2 | ZINC02782238 | ZINC02782238 | <rdkit.Chem.rdchem.Mol object at 0xf4e8698c0> |
| 3 | ZINC67640948 | ZINC67640948 | <rdkit.Chem.rdchem.Mol object at 0xf4e869af0> |
| 4 | ZINC67756512 | ZINC67756512 | <rdkit.Chem.rdchem.Mol object at 0xf4e869f50> |
dataframe['cluster'] = np.nan
for indice in range(len(mlx_res['NewClusters'])):
for registro in mlx_res['NewClusters'][indice]:
dataframe.loc[registro, 'cluster'] = indice
dataframe.head(5)
| Name | ID | ROMol | cluster | |
|---|---|---|---|---|
| 0 | ZINC50764925 | ZINC50764925 | <rdkit.Chem.rdchem.Mol object at 0xcb08486d0> | 31.0 |
| 1 | ZINC65292537 | ZINC65292537 | <rdkit.Chem.rdchem.Mol object at 0xf4e8694d0> | 168.0 |
| 2 | ZINC02782238 | ZINC02782238 | <rdkit.Chem.rdchem.Mol object at 0xf4e8698c0> | 5661.0 |
| 3 | ZINC67640948 | ZINC67640948 | <rdkit.Chem.rdchem.Mol object at 0xf4e869af0> | 2058.0 |
| 4 | ZINC67756512 | ZINC67756512 | <rdkit.Chem.rdchem.Mol object at 0xf4e869f50> | 2283.0 |
PandasTools.WriteSDF(dataframe, 'output.sdf', molColName='ROMol', idName=None, properties=list(dataframe.columns), allNumeric=False, forceV3000=False)
The first part imports the libraries and then imports a random 50K molecules taken from ZINC. Then we generate the fingerprints that are needed for both the standard RDKit clustering and the MLX accelerated.
The first run used the RDKit clustering, the results are
--- RDKit (BulkTanimotoSimilarity + ClusterData) --- Similarity: 179.870s Clustering: 55.766s Total: 235.636s -> 11267 clusters (largest: 188)
I then ran the MLX accelerated clustering and the results are
--- MLX/Metal (Fused Tanimoto→CSR + Butina CPU) --- Fused sim→CSR: 5.589s (Metal, no N×N matrix) Butina: 0.253s (CPU CSR greedy) Total: 5.842s -> 11213 clusters (largest: 188) Edges: 809,760 | Memory saved: 10000 MB (no sim matrix)
This is a 40-fold improvement in time taken!! Clearly a great piece of work by the author.
The last part simply annotates each molecule with the appropriate cluster number and then exports the results to an sdf file.
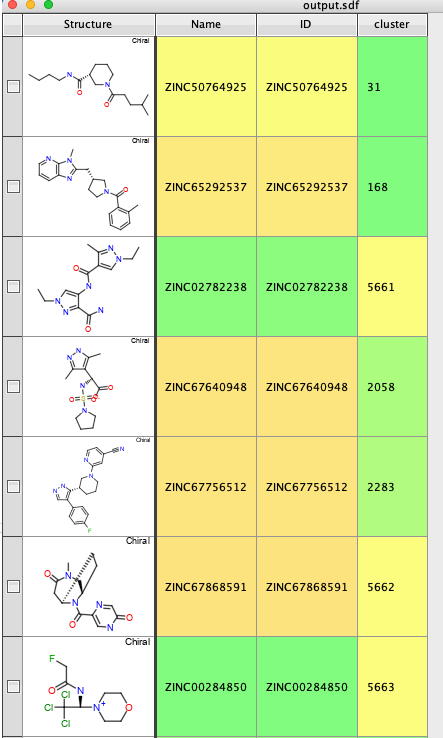
I tried clustering 150K molecules and got the following error.
--- MLX/Metal (Fused Tanimoto→CSR + Butina CPU) ---
libc++abi: terminating due to uncaught exception of type std::runtime_error: [METAL] Command buffer execution failed: Impacting Interactivity (0000000e:kIOGPUCommandBufferCallbackErrorImpactingInteractivity)
Unfortunately solving this is beyond my capabilities, but I've posted the issue on the repo.