In 2020 as a result of lockdown I was asked to help create a course for MRes students as an introduction to computer-aided drug design. [DOI]
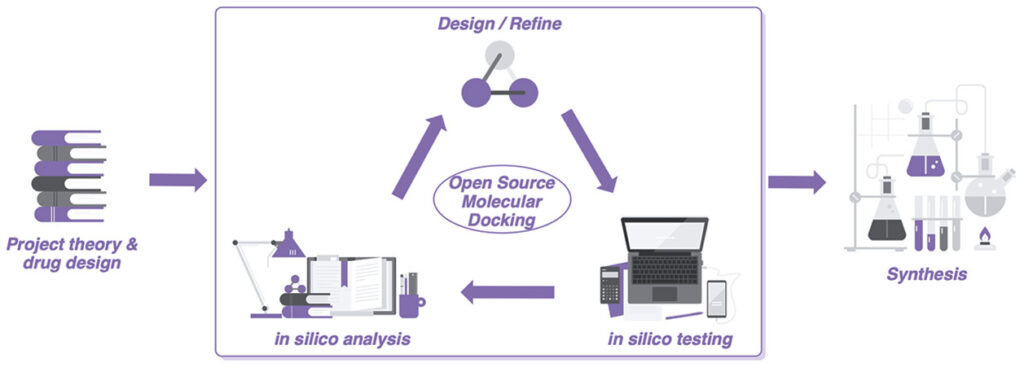
This work lead to the above publication, a lab manual and a Jupyter notebook all of which are freely available on GitHub
https://github.com/UCL/Open_Docking_Lab_Handbook
The Jupyter notebook has had a few updates and I thought I’d share the updated version but also the slide deck that accompanied the course introduction.
A Jupyter Notebook to aid Docking to protein¶
This notebook implements a typical protocol for docking ligands to a target protein. It uses RDKit (http://www.rdkit.org) to generate a number of reasonable conformations for each ligand and then uses SMINA (https://sourceforge.net/projects/smina/) to do the docking. Two methods of docking are implemented, the first docks into a rigid receptor, the second sets the protein side-chains around the active site to be flexible. Bear in mind flexible docking will be much, much slower.
In this notebook we will be docking ligands in the COVID Mpro.
import sys
from collections import defaultdict
import numpy as np
from rdkit import Chem
from rdkit.Chem import AllChem
from rdkit.Chem.Draw import IPythonConsole
from rdkit.Chem import PandasTools
import pandas as pd
import py3Dmol
IPythonConsole.ipython_3d=True
%matplotlib inline
import matplotlib
import matplotlib.pyplot as plt
File location of structures for docking and file format¶
First we need get the location of the input file of structures you want to dock, replace "Fordocking.sdf" with your file. You may want to rename the output file for conformations, and the output file containing the docked structures.
The sdf file needs to have the name included in the first line of each molecule record.
AEM 10028511
MOE2019 2D
22 24 0 0 0 0 0 0 0 0999 V2000 7.2040 -6.7290 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0 6.3790 -6.7290 0.0000 N 0 0 0 0 0 0 0 0 0 0 0 0
# File locations
sdfFilePath = 'Fordocking.sdf' # The input file of structures to generate conformations from
ConfoutputFilePath = 'ForDockingConfs.sdf' # Output file containing conformations for docking
inputMols = [x for x in Chem.SDMolSupplier(sdfFilePath,removeHs=False)]
# Assign atomic chirality based on the structures:
len(inputMols) # Check how many strucures
3
#Check that all molecules have a name
for i, mol in enumerate(inputMols):
if mol is None:
print('Warning: Failed to read molecule %s in %s' % (i, sdfFilePath))
if not mol.GetProp('_Name'):
print('Warning: No name for molecule %s in %s' % (i, sdfFilePath))
#option to view individual structures (comment out with # if not needed)
mol = inputMols[2]
mol
You appear to be running in JupyterLab (or JavaScript failed to load for some other reason). You need to install the 3dmol extension:
jupyter labextension install jupyterlab_3dmol
#View as grid
Chem.Draw.MolsToGridImage(inputMols,legends=[mol.GetProp('_Name') for mol in inputMols])

Conformation generation¶
From RDKit documentation. Disclaimer/Warning: Conformer generation is a difficult and subtle task.
The docking program SMINA will rotate around torsions which may be enough for some molecules, however it will not flip rings and probably not identify cis amides. For some molecules you may not need to do conformation generation (set numConfs = 1).
#edit numConfs to desired number
with Chem.SDWriter(ConfoutputFilePath) as w:
for mol in inputMols:
m = Chem.AddHs(mol)
cids = AllChem.EmbedMultipleConfs(m, numConfs=3, numThreads=0) #edit num confs
confs = m.GetConformers()
for c in confs:
w.write(m, confId=c.GetId())
ms = [x for x in Chem.SDMolSupplier(ConfoutputFilePath,removeHs=False)]
# Assign atomic chirality based on the structures:
for m in ms: Chem.AssignAtomChiralTagsFromStructure(m)
len(ms) # check how many conformations
9
Docking to Protein¶
After generating the conformations we can now do the docking. In this example we use smina which can be downloaded from https://sourceforge.net/projects/smina/ you will need to know where smina has been installed and edit the path if needed.
The coronavirus, known as severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), is the agent responsible for the 2019–2020 viral pneumonia outbreak of coronavirus disease 2019 (COVID-19). The main prortease Mpro is essential for cleaving the viral protein into the component enzymes and structral proteins and is thus essential for viral replication (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9283855/). This makes it an attractive drug target. At the strat of the pandemic the crytalised protein was subjected to a fragment screen at Diamond.(https://www.diamond.ac.uk/industry/Case-Studies/Case-study-Fragment-Screening-to-fight-COVID-19.html). The results were then made publically avaiable, and one of these structures was downloaded nd used for this study.
The image below shows the active site with a fragment bound, this is taken from the fragalysis webite (https://fragalysis.diamond.ac.uk/viewer/react/landing) and the file is Mpro-x0107_0.pdb
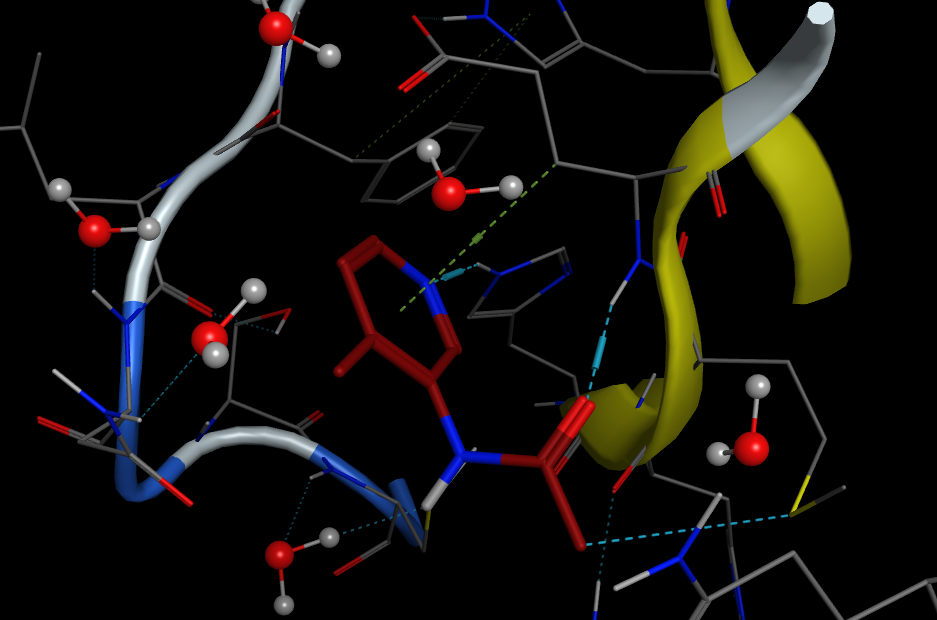
The file Mpro-x0107_0.pdb is in the folder containing the Jupyter notebook and you can view it in PyMOL. You can explore the binding site and note potential interactions.

Docking using smina
Need protein minus the ligand in pdb format,
the ligand extracted from binding site in pdb format,
Conformations to be docked as sdf from conformation generation above
DockedFilePath = ‘My_Docked.sdf’ is the File containing the Docked structures
ProteinForDocking = 'proteinNoligand.pdb'
LigandFromProtein = 'LigandOnly.pdb'
DockedFilePath = 'My_Docked.sdf'
FlexibleDockedFilePath = 'FlexDocked.sdf.gz'
!'/usr/local/smina.osx.12' --exhaustiveness 20 --cpu 10 --seed 0 --autobox_ligand '{LigandFromProtein}' -r '{ProteinForDocking}' -l '{ConfoutputFilePath}' -o '{DockedFilePath}'
_______ _______ _________ _ _______
( ____ \( )\__ __/( ( /|( ___ )
| ( \/| () () | ) ( | \ ( || ( ) |
| (_____ | || || | | | | \ | || (___) |
(_____ )| |(_)| | | | | (\ \) || ___ |
) || | | | | | | | \ || ( ) |
/\____) || ) ( |___) (___| ) \ || ) ( |
\_______)|/ \|\_______/|/ )_)|/ \|
smina is based off AutoDock Vina. Please cite appropriately.
Weights Terms
-0.035579 gauss(o=0,_w=0.5,_c=8)
-0.005156 gauss(o=3,_w=2,_c=8)
0.840245 repulsion(o=0,_c=8)
-0.035069 hydrophobic(g=0.5,_b=1.5,_c=8)
-0.587439 non_dir_h_bond(g=-0.7,_b=0,_c=8)
1.923 num_tors_div
Using random seed: 0
0% 10 20 30 40 50 60 70 80 90 100%
|----|----|----|----|----|----|----|----|----|----|
***************************************************
mode | affinity | dist from best mode
| (kcal/mol) | rmsd l.b.| rmsd u.b.
-----+------------+----------+----------
1 -5.2 0.000 0.000
2 -5.1 1.878 4.204
3 -4.9 2.059 2.089
4 -4.8 3.414 4.509
5 -4.8 3.800 5.196
6 -4.7 3.889 4.994
7 -4.7 1.792 4.236
8 -4.7 2.259 4.832
9 -4.6 2.004 2.862
Refine time 0.816
Using random seed: 0
0% 10 20 30 40 50 60 70 80 90 100%
|----|----|----|----|----|----|----|----|----|----|
***************************************************
mode | affinity | dist from best mode
| (kcal/mol) | rmsd l.b.| rmsd u.b.
-----+------------+----------+----------
1 -5.3 0.000 0.000
2 -5.2 1.902 4.360
3 -4.9 2.089 2.125
4 -4.8 1.840 4.368
5 -4.8 2.128 3.013
6 -4.8 3.455 4.611
7 -4.8 3.894 5.353
8 -4.7 2.311 4.931
9 -4.7 3.276 3.897
Refine time 0.828
Using random seed: 0
0% 10 20 30 40 50 60 70 80 90 100%
|----|----|----|----|----|----|----|----|----|----|
***************************************************
mode | affinity | dist from best mode
| (kcal/mol) | rmsd l.b.| rmsd u.b.
-----+------------+----------+----------
1 -5.2 0.000 0.000
2 -5.1 1.856 4.214
3 -5.1 1.501 1.547
4 -4.8 3.404 4.512
5 -4.7 2.263 4.837
6 -4.7 3.854 4.963
7 -4.6 2.077 2.920
8 -4.6 1.776 4.252
9 -4.6 1.594 2.479
Refine time 0.827
Using random seed: 0
0% 10 20 30 40 50 60 70 80 90 100%
|----|----|----|----|----|----|----|----|----|----|
***************************************************
mode | affinity | dist from best mode
| (kcal/mol) | rmsd l.b.| rmsd u.b.
-----+------------+----------+----------
1 -6.1 0.000 0.000
2 -5.6 1.134 1.807
3 -5.5 1.552 6.351
4 -5.4 1.620 6.086
5 -5.3 3.583 6.277
6 -5.3 3.413 4.377
7 -5.2 3.492 4.620
8 -4.9 3.171 4.376
9 -4.9 4.155 5.944
Refine time 2.101
Using random seed: 0
0% 10 20 30 40 50 60 70 80 90 100%
|----|----|----|----|----|----|----|----|----|----|
***************************************************
mode | affinity | dist from best mode
| (kcal/mol) | rmsd l.b.| rmsd u.b.
-----+------------+----------+----------
1 -6.1 0.000 0.000
2 -5.6 1.128 1.791
3 -5.5 1.445 6.249
4 -5.4 3.569 6.286
5 -5.3 1.605 6.169
6 -5.2 1.787 6.309
7 -5.2 3.426 4.599
8 -5.1 3.386 4.568
9 -5.0 4.180 5.916
Refine time 2.062
Using random seed: 0
0% 10 20 30 40 50 60 70 80 90 100%
|----|----|----|----|----|----|----|----|----|----|
***************************************************
mode | affinity | dist from best mode
| (kcal/mol) | rmsd l.b.| rmsd u.b.
-----+------------+----------+----------
1 -6.1 0.000 0.000
2 -5.5 1.136 1.787
3 -5.5 3.686 6.643
4 -5.5 3.505 4.463
5 -5.3 1.483 6.278
6 -5.3 2.067 6.362
7 -5.2 1.603 6.200
8 -5.2 1.586 6.120
9 -5.2 1.570 1.637
Refine time 2.101
Using random seed: 0
0% 10 20 30 40 50 60 70 80 90 100%
|----|----|----|----|----|----|----|----|----|----|
***************************************************
mode | affinity | dist from best mode
| (kcal/mol) | rmsd l.b.| rmsd u.b.
-----+------------+----------+----------
1 -6.3 0.000 0.000
2 -5.6 1.417 6.083
3 -5.4 1.670 6.325
4 -5.4 1.045 1.706
5 -5.3 1.672 6.123
6 -5.0 4.176 5.478
7 -4.6 3.917 5.291
8 -4.6 3.172 4.801
9 -4.6 3.207 4.829
Refine time 2.248
Using random seed: 0
0% 10 20 30 40 50 60 70 80 90 100%
|----|----|----|----|----|----|----|----|----|----|
***************************************************
mode | affinity | dist from best mode
| (kcal/mol) | rmsd l.b.| rmsd u.b.
-----+------------+----------+----------
1 -6.4 0.000 0.000
2 -5.9 2.402 4.336
3 -5.9 1.442 2.028
4 -5.6 1.625 4.578
5 -5.6 1.832 4.599
6 -5.6 2.390 4.478
7 -5.5 1.425 2.031
8 -5.5 1.877 4.547
9 -5.4 1.960 2.951
Refine time 1.710
Using random seed: 0
0% 10 20 30 40 50 60 70 80 90 100%
|----|----|----|----|----|----|----|----|----|----|
***************************************************
mode | affinity | dist from best mode
| (kcal/mol) | rmsd l.b.| rmsd u.b.
-----+------------+----------+----------
1 -6.2 0.000 0.000
2 -5.7 1.385 6.033
3 -5.7 1.158 1.826
4 -5.3 1.068 1.735
5 -5.3 1.657 6.172
6 -5.0 4.262 5.523
7 -5.0 1.365 5.916
8 -4.8 3.164 4.922
9 -4.8 3.953 5.164
Refine time 2.121
Loop time 15.729
Flexible docking method, set all side chains within specified distance to flexdist_ligand to flexible This will take an order of magnitude longer. Currently disabled, to enable remove the # Then add the list of residues you want to flex by editing –flexres ***
#!'/usr/local/bin/smina.osx.12' --cpu 10 --seed 0 --autobox_ligand '{LigandFromProtein}' --autobox_add 5 -r '{ProteinForDocking}' --flexres *** -l '{ConfoutputFilePath}' -o '{FlexibleDockedFilePath}'
#View results
#this a way to quickly scan through the results to check
import gzip
v = py3Dmol.view()
v.addModel(open('proteinNoligand.pdb').read())
v.setStyle({'cartoon':{},'stick':{'radius':.1}})
v.addModel(open('LigandOnly.pdb').read())
v.setStyle({'model':1},{'stick':{'colorscheme':'redCarbon','radius':.125}})
#v.addModelsAsFrames(gziopen('My_Docked.sdf.gz','rt').read())
v.addModelsAsFrames(open('My_Docked.sdf','rt').read())
v.setStyle({'model':2},{'stick':{'colorscheme':'greenCarbon'}})
v.animate({'interval':1000})
v.zoomTo({'model':2})
v.rotate(90)
You appear to be running in JupyterLab (or JavaScript failed to load for some other reason). You need to install the 3dmol extension:
jupyter labextension install jupyterlab_3dmol
The slide deck is also included, this provides a link to the lab manual that can be downloaded from GitHub

The folder containing notebook, slide deck, and associated file can all be downloaded here.
The lab manual and the original notebook can be found here
https://github.com/UCL/Open_Docking_Lab_Handbook
If this is useful please cite the orginal publication
Molecular Docking with Open Access Software: Development of an Online Laboratory Handbook and Remote Workflow for Chemistry and Pharmacy Master’s Students to Undertake Computer-Aided Drug Design, Bethanie A. Clent, Yuhang Wang, Hugh C. Britton, Frank Otto, Christopher J. Swain, Matthew H. Todd, Jonathan D. Wilden, Alethea B. Tabor. J. Chem. Educ. 2021, 98, 9, 2899–2905 [DOI]
Updated
As might be expected during the course the students asked many interesting questions and I attempted to answer them with additional webinars on topics such as File Types, Molecular Interactions, Creating schematics of poses etc. These slide decks are not particularly logically organised since they were created in response to questions but I’ve now uploaded these to the server as well and they can be downloaded here,
