It is sometimes difficult to keep the website up to date with some of the software that I don’t use and so I was delighted when Fedor Goumans contacted me to highlight some of the updates at Scientific Computing and Modelling. While ADF is still SCM’s most widely used program, SCM offers a comprehensive modelling suite consisting of several different tools. There is also a GUI that allows users access to all the different programs it also includes a structure builder that is particularly useful for constructing periodic systems.
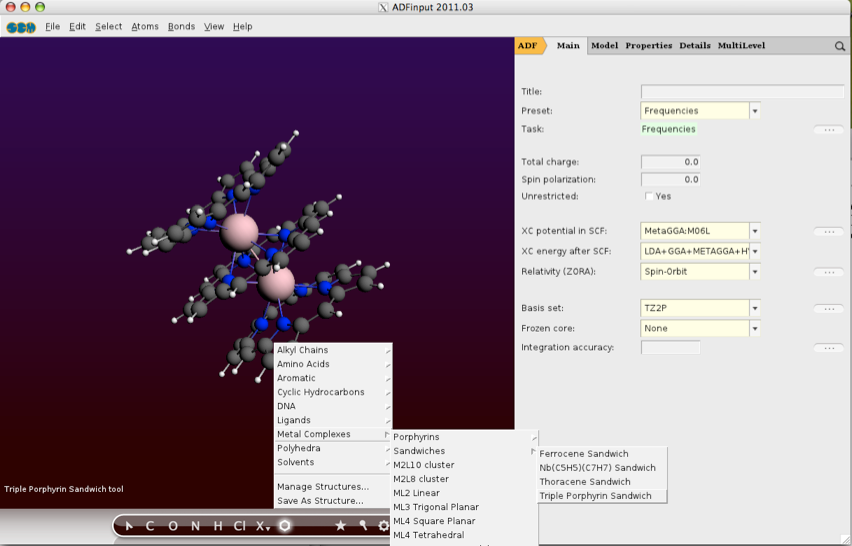
ADF which is updated regularly, is a powerful computational chemistry program designed to understand and predict chemical structure and reactivity using density functional theory (DFT), in particular heavy elements are accurately modelled using ZORA.
A number of spectroscopic properties can be calculated in ADF
- Vibrational spectroscopy
- Infrared (IR) frequencies and intensities
- VCD, MBH, Franck-Condon factors
- (resonance) Raman, VROA
- Electronic spectroscopy (with time-dependent DFT)
- UV/Vis
- X-ray: NEXAFS, XANES
- Large systems: coupled FDE, state-selective excitations
- frequency-dependent (hyper-)polarizability (NLO) lifetime effects, dispersion coefficients
- circular dichroism (CD), optical rotation (ORD)
- magnetizibility, MCD, Verdet constant
- Nuclear Magnetic Resonance (NMR) spectroscopy
- chemical shift, spin-spin coupling, paramagnetic NMR
- Electron paramagnetic resonance (EPR/ESR) spectroscopy
- EPR g-tensor, hyperfine interaction (A-tensor), ZFS
- Nuclear quadropole interaction (EFG), EPR Q-tensor
- Mössbauer spectroscopy, NRVS
BAND is the periodic density functional theory extension of the molecular code ADF. Localised atomic orbital (LCAO) basis sets allow for the proper modeling of 1D (polymers) and 2D (surfaces) periodic structures without artefacts and reduced performance. There are many examples of the use of BAND hereincluding examination of the surface of metal alloys and the structure and reactivity of crystals.
DFTB provides accurate results at a fraction of the cost of a DFT evaluation through parameterization of the integrals. Long-range interactions are described with empirical dispersion corrections and the novel DFTB3 approach handles charged systems accurately.
ReaxFF is a program for modeling chemical reaction dynamics with atomistic potentials based on the reactive force field approach developed by van Duin and coworkers. SCM has implemented and parallelized ReaxFF and significantly optimized the original code, removing memory bottlenecks. Systems consisting of a 3D box of multiple molecules totaling hundreds of thousands of atoms can now be modeled on a modern desktop computer. ReaxFF has been used to study crystal growth and dissolution, catalytic surfaces, and combustion chemistry.
COSMO-RS (COnductor like Screening MOdel for Realistic Solvents) is designed to predict the properties of fluids and solutions including
- activity coefficients, solvation free energies, Henry’s law constants
- solubilities
- partition coefficients (log P, log kOW)
- vapor pressure, boiling point of a solvent or mixture
- vapor-liquid diagrams binary and ternary mixtures (VLE/LLE)
- excess energies, azeotropes, miscibility gaps
- composition lines, flash points
- pKa values
SCM also distributes MOPAC2012 this is useful for modeling basic properties of molecules more approximately but more quickly than density functional theory, and are often used to set up a geometry optimisation to accelerate a subsequent ADF calculation. The GUI allows users to switch between MOPAC2012 and the other programs described above.
Updated 20 August 2013