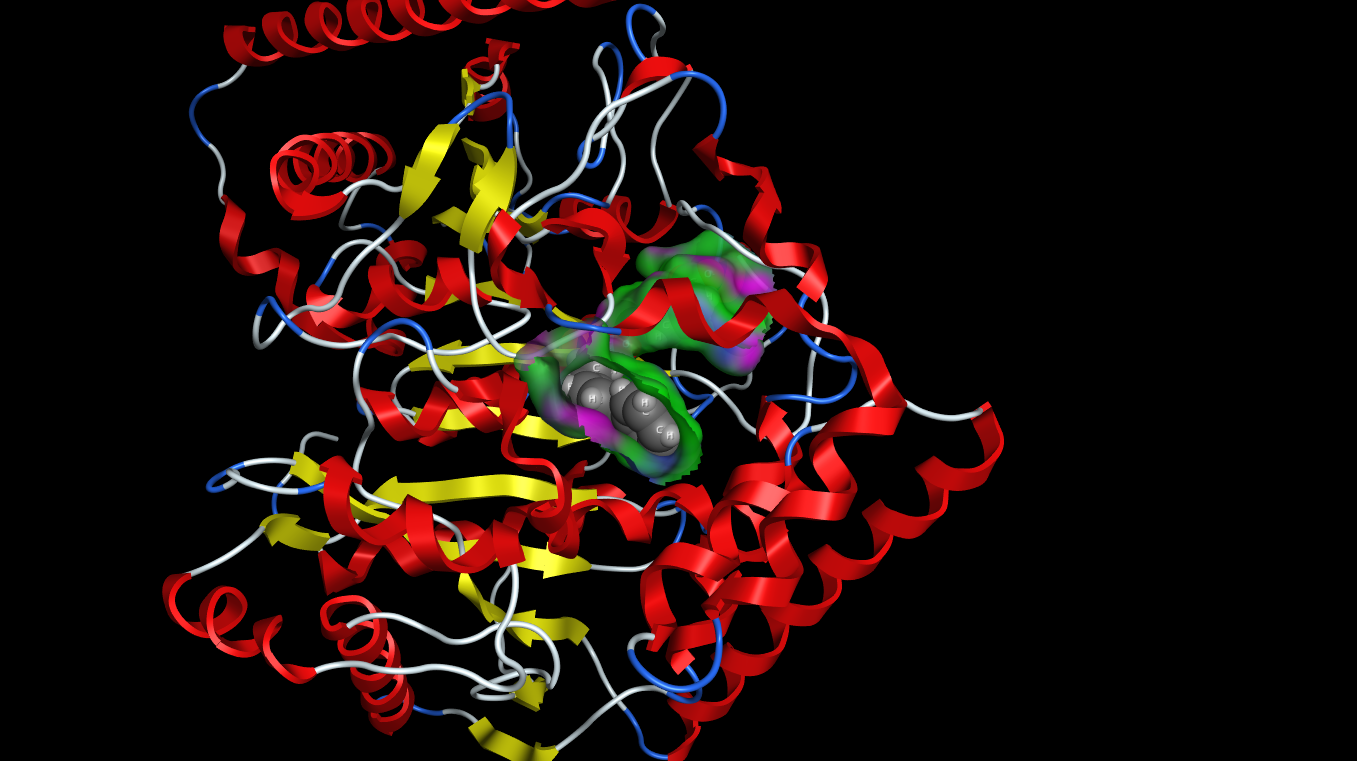
I’ve been using the latest update to MOE from Chemical Computing Group for a while now and i thought I’d post a few impressions. Perhaps the most striking update is the graphics, the 3D graphics capabilities in MOE have been substantially updated to make use of modern Graphics Processing Units. In addition to shadows the updated graphics are now similar in quality to raytracing.
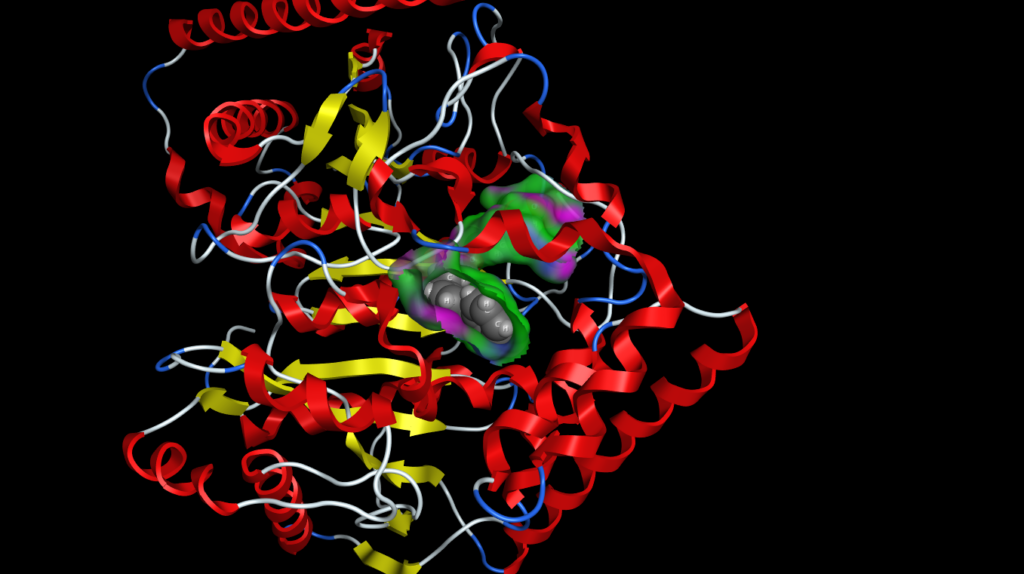
Surfaces, fonts and atoms are now rendered beautifully and even fonts are true 3D objects and fonts further away are scaled down to look smaller. The atom and ribbon rendering style menu items have now been collected into a dedicated panel. This had me hunting for missing menu items intially but now I’m used to it and it allows you to change several options in one panel rather than selecting several menu items. These panels can also be accessed by right-clicking in the MOE main window, or clicking on the buttons at the bottom right on the manin MOE window. The same panels can also be accessed from the Sequence Editor window allowing easy manipulation of protein ligand complexes.

There are a couple of new functions.
Scaffold Replacement
A new application for small molecule Scaffold Replacement, Fragment Linking and Ligand Growth has been added. The methodology uses Pharmacophore Search queries to search a 3D fragment database for molecules that satisfy R-group bond vector constraints. The application performs the following:-
Automatic Query Generation. From an input 3D small molecule, possibly in a receptor pocket, Pharmacophore Search queries using the Link features are automatically generated based upon user-specified R-groups. 3D Search. A 3D database is searched for fragments that can add an R-group to a molecule, link separate fragments, replace a scaffold. Cyclization is support as well as multiple-bond formation. The complete molecules are formed with dihedral angle and clash tests. 2D and 3D Filters. A set of descriptor filters can be applied during the search as well as any QuaSAR Model files and/or additional pharmacophore filters (shape, interaction requirements, etc.). Refinement and Scoring. The coordinates of the constructed molecules can be automatically refined possibly in the context of an active site. MM/GBVI or other docking scoring functions can be applied. Synthetic feasibility and stability scores are automatically calculated.
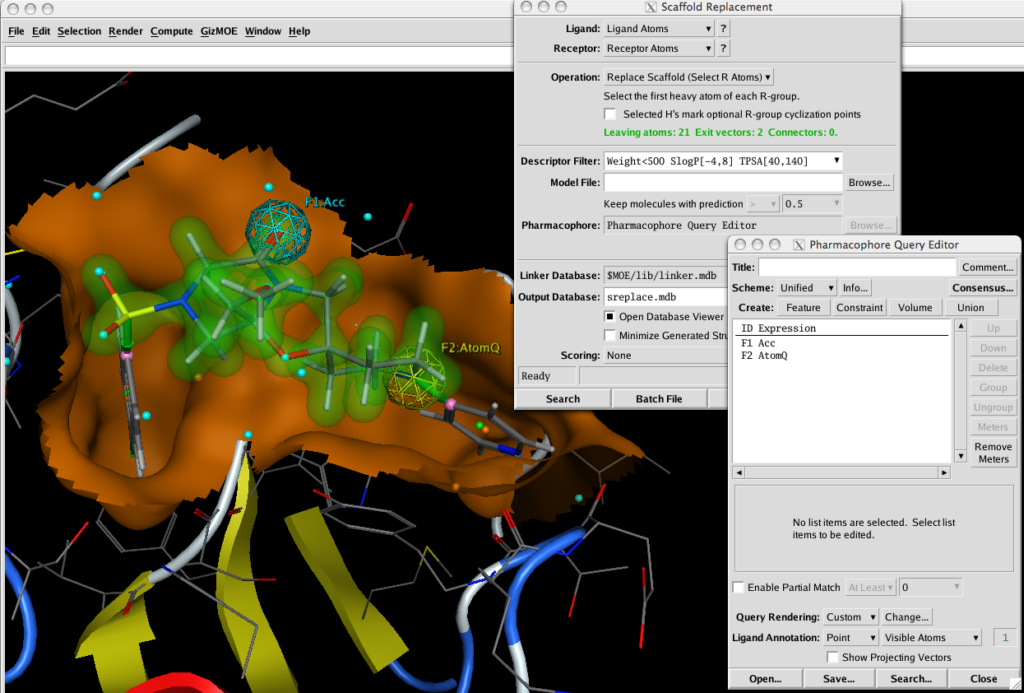
With Scaffold Replacement Panel open selecting two atoms highlights the intervening atoms as the scaffold and creates two exit vectors (green). You can simply run the query now, or include pharmacophoric elements to refine the results. Using the Pharmacophore Query Editor it is trivial to select atoms and create pharmacophore features.
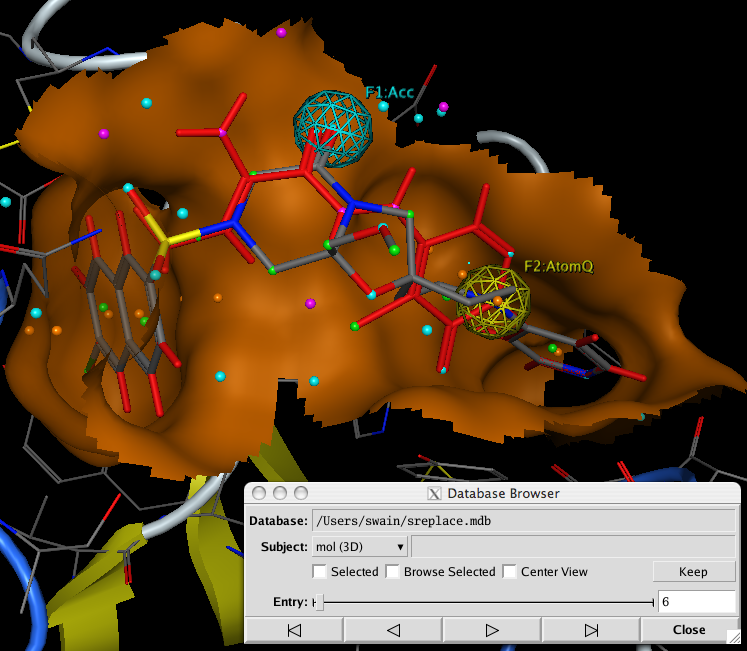
After running the query a database of results is created, using the Database Browser allows you to rapidly scan through the results, with the molecules created docked into the active site. It is of course possible to further refine the results by minimising the structures within the active site. Or using the new GOLD interface.
Synthetic Feasibility Score
A new algorithm for assessing the chemical feasibility or plausibility of a molecule has been added to MOE (Synthetic Feasibility Score). The algorithm performs repeated bond disconnections according to retrosynthetic rules until no more disconnections can be made. Each disconnection fragments the molecule into one or more further fragments each of which is a molecule. For example, in ester clevage reactions, hydroxyl groups are added to “cap” the fragments. Each resulting molecule fragment resulting from the disconnection is converted to a unique SMILES representation and a lookup is conducted in a “starting materials” database (in $MOE/lib). The starting materials database was produced from a large collection of molecules subjected to the disconnection procedure. A score is derived for each heavy atom in the original molecule. If an original atom ended up in a fragment that was found in the starting materials database, then the atom is considered covered. The score for the whole molecule is the percentage of covered heavy atoms of in the original molecule. So while the algorithm highlights compounds that might be difficult to make it does not really distinguish between those accesible via either very long or short synthetic routes, or compounds that might be accessed via a common route.
SOAP Server. A new executable has been added to MOE that implements SOAP server functionality for use in web services programming, or used to add cheminformatics funtionality to other applications. The program can keeps a collection of moebatch processes running and instructs them to perform calculations as web requests are received. The moeweb server allows SVL functions to be exposed as web services using the SOAP/WSDL protocols. The standard functionality includes calculating medicinal chemistry properties, 2D depiction layout and SMILES to MDL MOL interconversion. I’ve not used this but plan to explore it in the future.
This is a useful update to MOE providing a much needed improvement to the graphics capabilities and general tidying of the menu layouts. The scaffold replacement utility is an invaluable addition to the structure-based design toolset. I’ve yet to try the new SOAP server, but I suspect in some organisations it may turn out to be very useful, particularly if you have built existing SVL tools.