LigandScout from inte:ligand is a software tool that allows to rapidly and transparently derive 3D pharmacophores from structural data of macromolecule/ligand complexes in a fully automated and convenient way. There is a free evaluation that can be downloaded from the website you just need to register to get a one month evaluation license. It runs under MacOSX, Windows and Linux and I should say from the outset that this feels very much like a proper Mac OS X application not a UNIX port. From the installer to the standard Mac OS X keyboard short cuts this has the look and feel of a Mac application.
After installation, on first start you are prompted for the serial number and then you are presented with the LigandScout desktop.
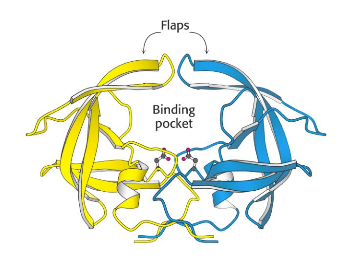
To review LigandScout I thought it might be useful to use the outstanding work by the DupontMerck group on HIV Protease (Lam, P.Y., Jadhav, P.K., Eyermann, C.J., Hodge, C.N., Ru, Y., Bacheler, L.T., Meek, J.L., Otto, M.J., Rayner, M.M., Wong, Y.N., et al. (1994) Rational design of potent, bioavailable, nonpeptide cyclic ureas as HIV protease inhibitors. Science 263: 380-384). HIV protease is an Aspartyl Proteinase that is formed as dimer. It is well validated target, the enzyme is unique to HIV virus and is not present in humans. It has been shown that active HIV protease is required for viral infectivity and a mutant HIV provirus (Asp to Asn) was unable to infect MT-4 lymphoids (Proc. Natl. Acad. Sci. USA, 85, 4686-490 July 1988. ). The C2-symmetric nature of the HIV protease homodimer provides a unique property of the enzyme that could be exploited in the drug design process. X-ray data revealed that both the N- and C-termini of the asymmetric substrates and the inhibitors bind to identical subsites. Each monomer provides one of the two essential aspartic acids.
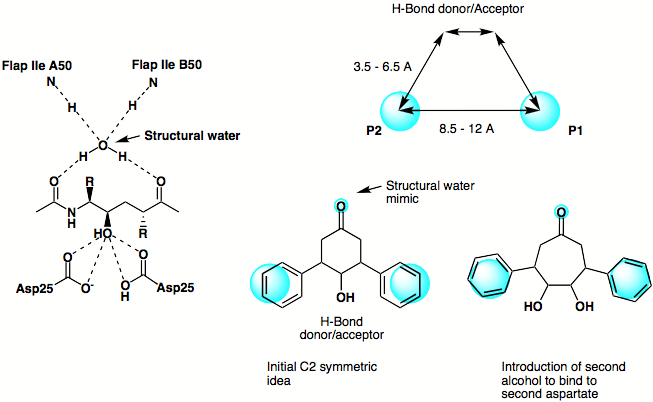
The original design phlosophy is shown below, based on the X-ray structure of a peptide based inhibitor bound in the active site of the protein a simple scematic pharmacophore was devised and series of simple C2 symmetric ligands devised. The seven membered ketone was replaced by the corresponding amide in order to enhance the putative hydrogen bonding interactions achieved by mimicing the structural water.
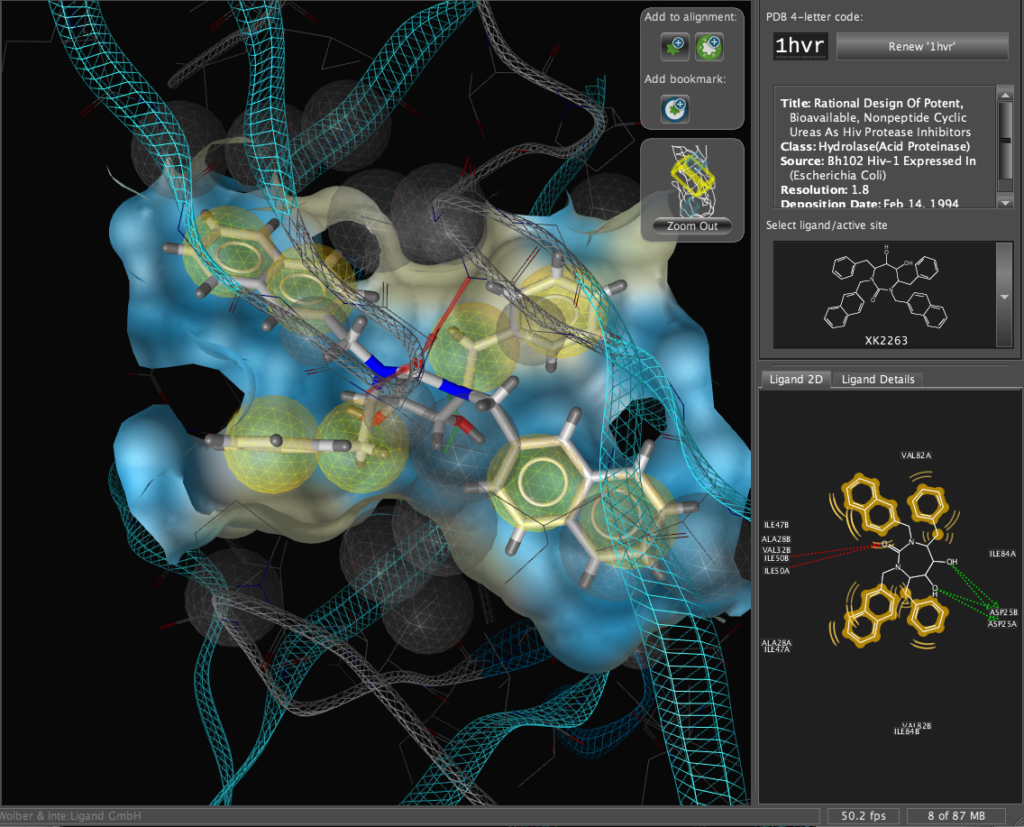
The X-ray structure of the resulting complexes have been published and are available for download from the Protein Data Bank, the code for the required structure is 1hvr. Simply type the 4-digit code into the PDB code box and click the “Renew ‘1hvr'” button.
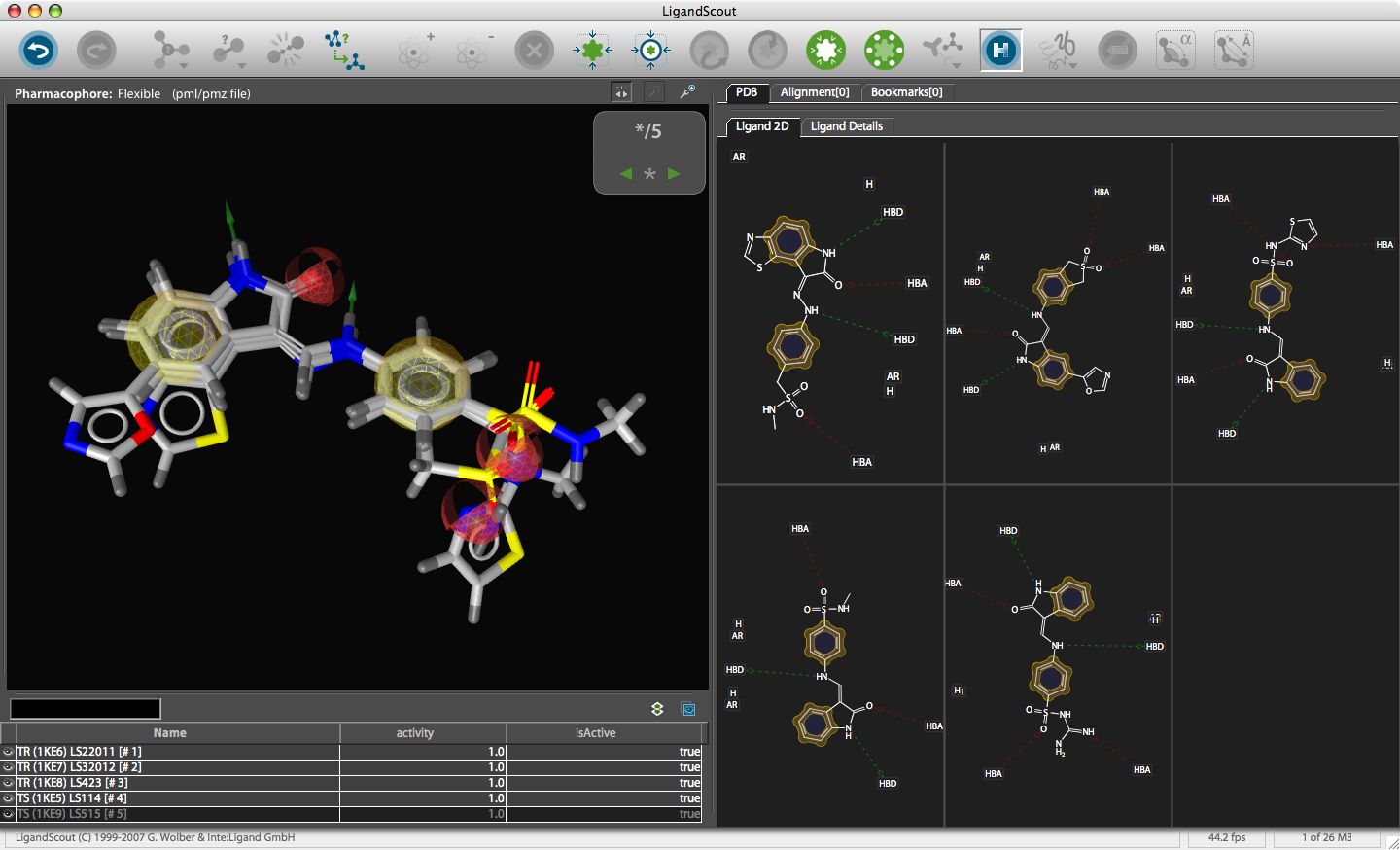
The corresponding file will be automatically downloaded and the automatic PDB interpretation algorithm initated, the resulting protein-ligand complex is then displayed in the Macromolecule View. The algorithm automatically assigns secondary structure, identifies ligands and then creates a colour-coded view of the complex, with ligands highlighted by yellow translucent boxes.
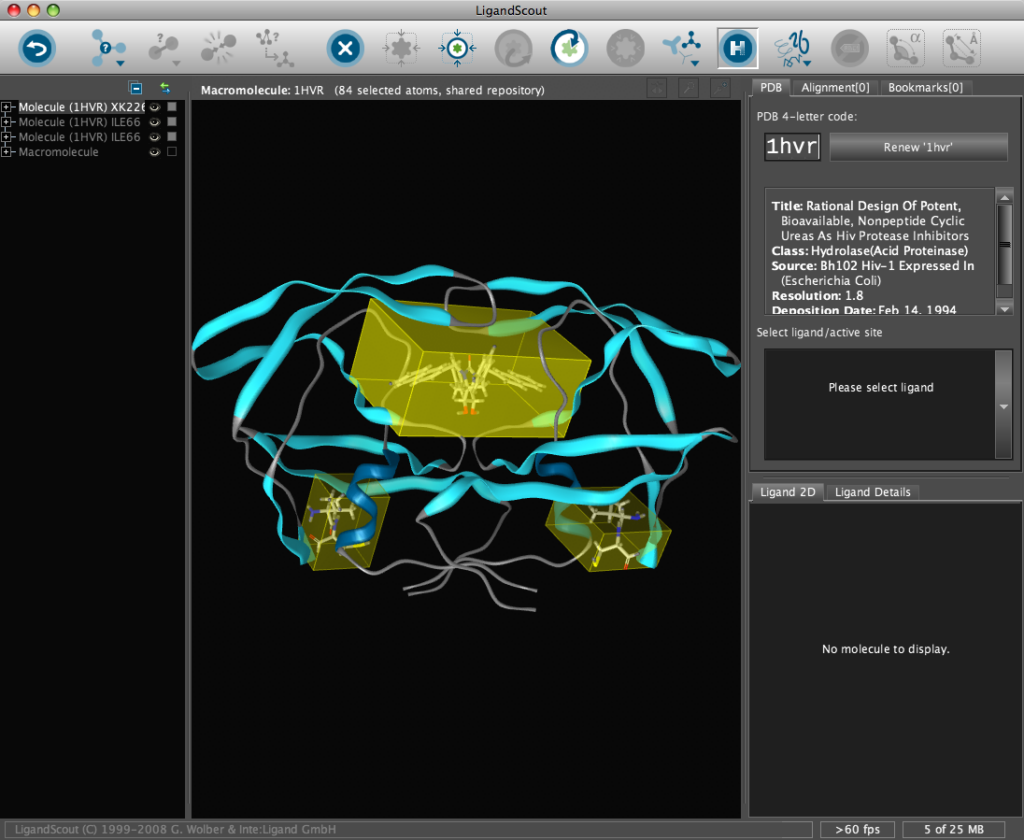
In addition all the components are fully described in the left hand hierarchy view panel allowing rapid access to viewing options for each element of the complex right down to individual atoms. So the user can toggle the display of an individual element very easily.If you run the mouse over the ligands a 2D representation fades into view at the bottom edge of the 3D Macromolecule View allowing you to quickly identify the drug ligand.
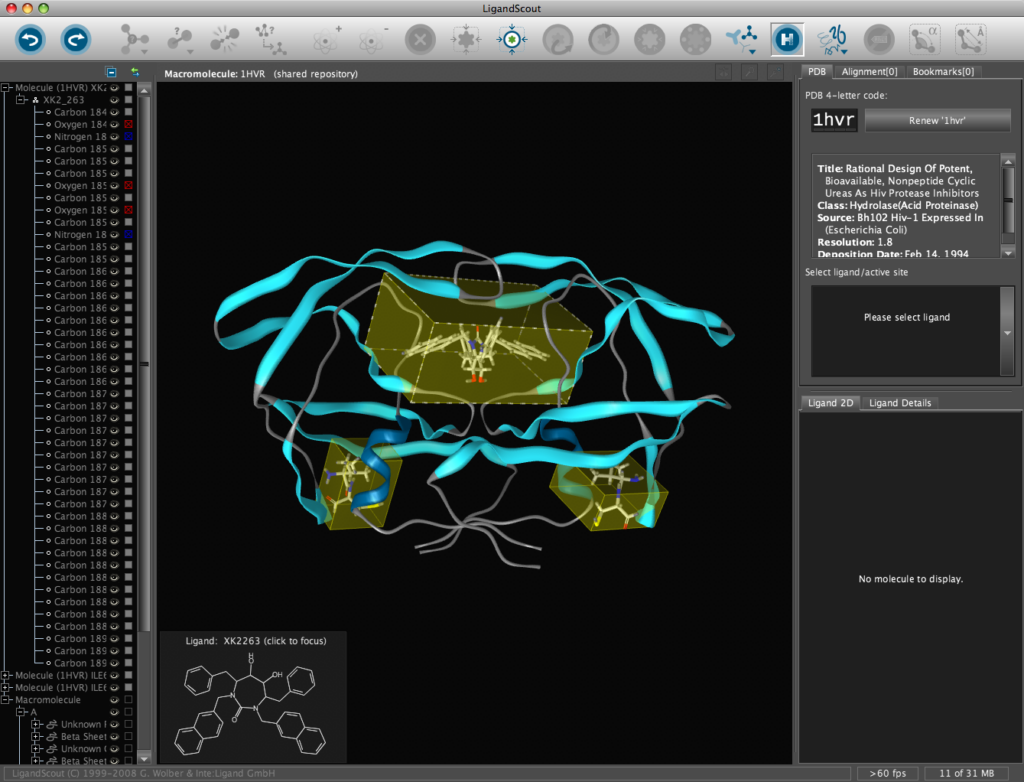
If you double-click on the ligand the focus of the display smoothly rotates and magnifies to put the ligand as the central focus. The Ligand 2D pane now displays a 2D representation of the ligand. The way that everything happens is beautifully orchestrated and really encourages the user to explore. LigandScout supports the scroll wheel on my two button mouse and the user can srcoll in and out, rotate or translate the display using the left and right buttons. The display is excellent with no evidence of flickering even with complex structures. LigandScout supports all the expected display functions for both ligand (stck, ball and stick, space-filling) and protein (stick, wireframe, secondary structure etc.).
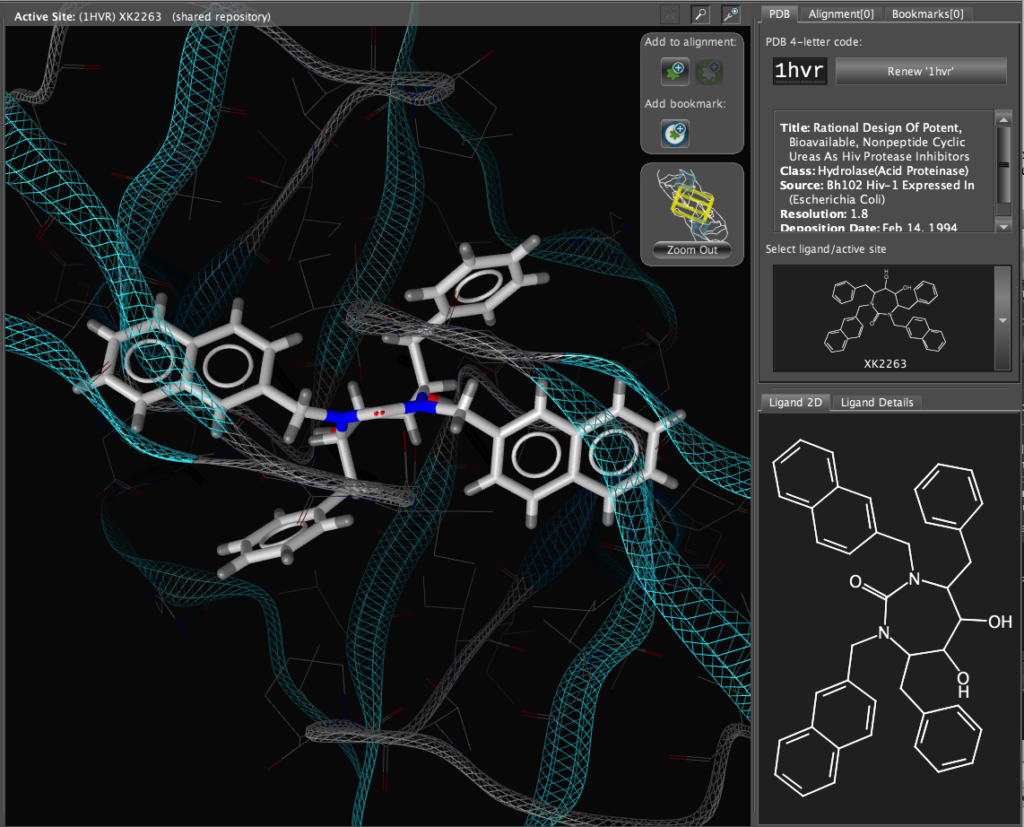
With the focus on the ligand it is possible to really take advantage of some of the display features. With a single click we can add a surface to the protein binding pocket, what is not easy to see from the static images is the way the frontmost suface fades away as the complex is rotated so that you can always see into the pocket, the surface automatically reappears when not obstructing the view. This dynamic updating is a fabulous aid to exploring the structure. The surface can be colour coded based on polarity, lipophilicity or charge (MMFF94).
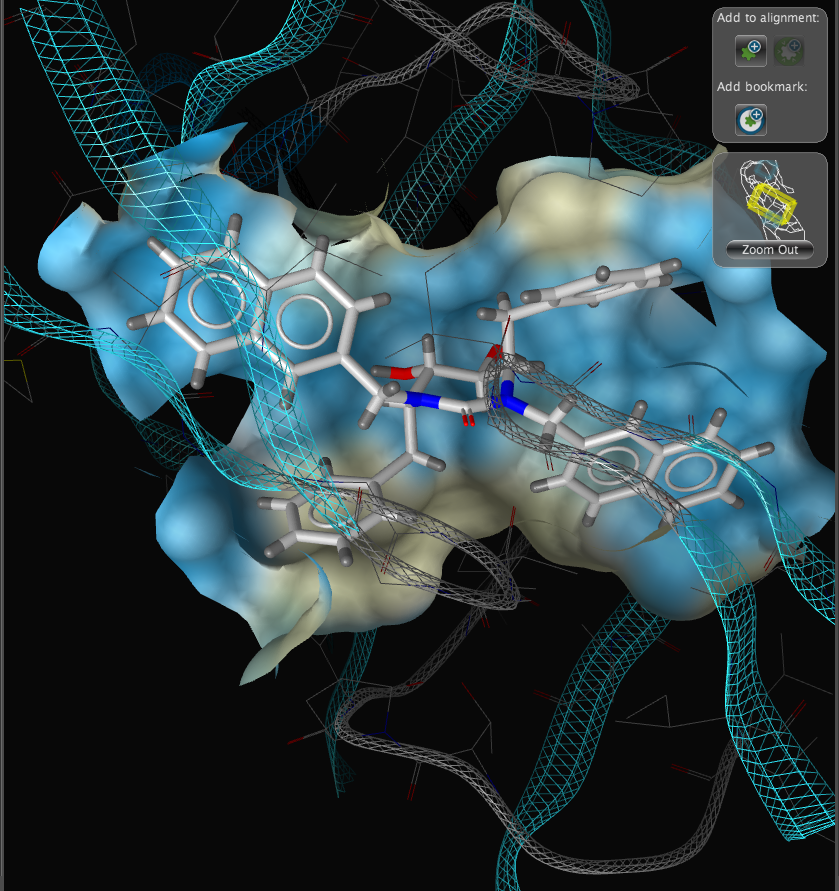
With another click you can automatically generate a pharmacophore, a “pharmacophore” is defined as a collection of chemical features that characterize a specific mode of interaction of a ligand in the active site of the macromolecule in 3D space. Chemical features are e.g. hydrogen bonds, charge interactions, hydrophobic areas but can also include unfavourable steric interactions. The phatmacophore can be used by a chemist in the optimisation of a potential drug ligand but because the pharmacophore can usually defined very simply it is ideal for use in computationally searching compound libraries. LigandScout supports the following pharmacophoric elements :-
- Hydrogen Bond Donor
- Hydrogen Bond Acceptor
- Positive Ionizable Area
- Negative Ionizable Area
- Hydrophobic Interactions
- Aromatic Ring
- Metal Binding Feature
- Excluded Volume
The results are displayed on both the 3D macromolecular complex view but also in the 2D ligand annotation. Clicking on an element in either the 2D or 3D display causes the element to be also highlighted in the other display.
If you are using the software-packages Catalyst, MOE or Phase it is recommended to use the Create Simplified Pharmacophore routines to achieve best compatibility to these external software applications since not all the screening applications use exactly the same definitions. You can use LigandScout to allign structures and pharmacophores by simply adding them to the alignment pane, then in the alignment pane selecting the desired structures and clicking on the alignment button. Here I show the alignment generated for two ligands from the X-ray structures 1hvr and 1hwr. inte:ligand also provide a couple of moviesshowing how this is done. If you have diverse structures binding to the same protein you can combine the pharmacophores and then select which features you want to include in a pharmacophore query search. LigandScout has some capability for ligand modification and minimisation but I would only use this for minor modifications, perhaps correcting tautomers etc.
Building a MOE Pharmacophore
Use the Create Simplified Pharmacophore (MOE) command in the Pharmacophore menu. In this case, LigandScout achieves compatibility by merging features at the same position and by adding charge features if and only if one of the underlying atoms have an explicit charge. Also the hydrogen bond lengths have to be adjusted to be 1.8 Å. Open the Save dialog in the File menu. Choose Pharmacophore in CCG MOE Format (*.ph4) in the file type drop-down menu, enter a file name and export the pharmacophore in one of the two supported MOE pharmacophore schemes. Also export the ligand as MDL .mol file for subsequent hypothesis verification. The *.ph4 can be used within the MOE environment to search databases of chemical structures. Whilst the results could be viewed in MOE the ease of use and the graphics in LigandScout are so good it might be attractive to return the hits to LigandScout for exploration, in particular the library view allows you to compare hoe different ligands fit in the pharmacophore easily.
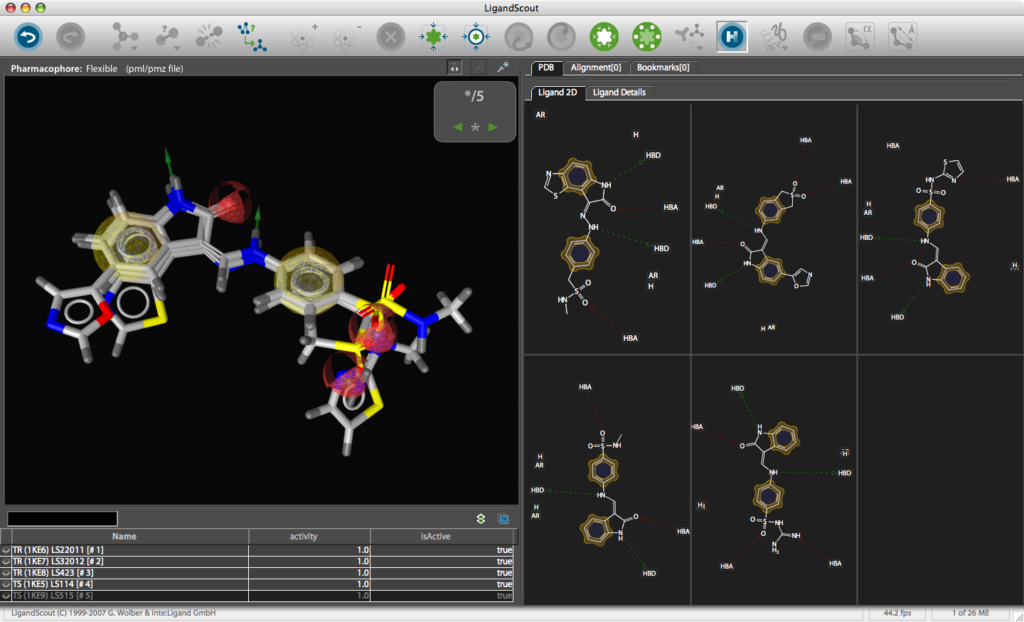
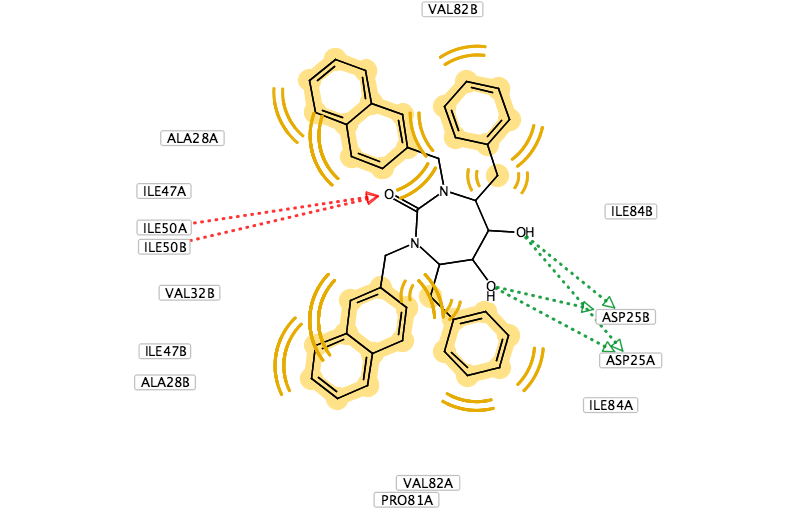
You can use LigandScout to prepare diagrams for publications and presentations, the 2D layout of the pharmacophore allows you to move the position of aminoacids to give a clearer picture and you can change the backgroud to white.
Summary
I have to say I’ve been really impressed with LigandScout, it is a fairly intuitive application and it really encourages the user to explore the structures and interactions. The program is sufficiently easy to use that academic of small companies without access to specialist computational chemists could use it profitably, in larger companies it would be invaluable tool to allow bench chemists to interact with specialist molecular modellers. I have a few minor quibbles, whilst LigandScout can create a MOE pharmacophore file that can be used for virtual screening you can’t save the molecule as the native MOE file format, also you can’t read native MOE files or multimolecule mdb databases. All small molecule structural information has to transferred as .mol, .sdf or mol2 format. It would also be nice to have an “Open recent items” option to quickly access previous work. It would also be useful to have tool tips (names that appear when the cursor hovers over a button) for the icons in the menu bar to aid new users.