High-throughput screening (HTS) remains a cornerstone of drug discovery, but searching through the many thousands of potential hits remains a daunting process. One aspect of judging whether a hit is genuine is to look at the activity of similar structures, based on the principle that similar structures are likely to have similar biological properties. Because of this key observation many different similarity measures and clustering techniques have been developed to aid analysis of HTS results.
Structural descriptor based methods are very commonly used, there are thousands of molecular descriptors available that can be used to provide a molecular fingerprint that can then be used for similarity scoring. These methods are computationally simple, rapid and generally effective, however they often don’t represent the “medicinal chemists” view of similarity. In contrast similarity measures based on maximum common substructure (MCS) usually do represent the chemists view. Most chemists would align structures based on a key structural framework or template and intrepret the influence of substituents on the template as initial structure-activity information. However computationally clustering using MCS is a major challenge due to the NP-complete nature to the problem. See J. Chem. Inf. Comput. Sci. 1998, 38, 915-924 for more details.
LibraryMCS is a tool from ChemAxon that uses hierachial clustering to sort molecules. Initial structures are found at the bottom of the hierarchy. The next level contains the maximum common structures of clusters of initial molecules, subsequent levels provide larger clusters of smaller commom substructures. The maximum common structures of a compound library can be searched by the libmcs a command line tool and I found it a useful way of investigating the underlying algorithm without using the interface which I found rather confusing to start with. In particular because there is no help currently implemented.
MacBookPro:~ username$ /Applications/ChemAxon/JChem/bin/libmcs -h
Library MCS - Maximum Common Substructure Clustering 0.7, (C) 2006-2008 ChemAxon Ltd.
Clusters input structure with respect to shared common substructures.
Usage: Library MCS [input file] [options]
Options:
-h, --help this help message
-v, --verbose progres monitoring and other messages
-e, --exact exact MCS recognition
-f, --fast fast, yet fairly accurate MCS recognition
-t, --turbo fastest and less reliable MCS recognition
-n, --minMCS integer value specifying the MCS size
where clustering terminates -m, --match (a|b|c|r) (+|-) turns matching contraints on (+), off (-)
for atom types (a), bond types (b), formak charges (c) and rings (r)
-o, --output SDF output file, terminal if -o omitted
-o, --output CSV CSV output file
-r, --report generate report (cluster statistics)
The brief help gives a insight into how the program works, the user can define -n the minMCS so that when set, for instance to 5, possible common structures smaller than 5 atoms are abandoned. You can use the -t (turbo) option to quickly explore a data set, or export the results as an sdf file or in SMILES as a csv file. Another option,¬†¬†-m, –match can be used to specify the conditions for considering two substructures common. By default, only identical substructures are common, that is, atom and bond types etc. should be the same in the two (or more) structures. This strict condition, however, can be relaxed by allowing the pairing of single, double and aromatic bonds, or different atoms types, charged and non charges atoms, sp2 and sp3 atoms etc. This way more generalised scaffolds can be obtained. It would be useful if it allowed the pairing of rings of different sizes, e.g. 5-membered aromatic ring can match a 6-membered one, since thiophene is a well known bioisostere of benzene.LibraryMCS is a graphical user interface to the application and to be honest it feels like a work in progress, whilst the underlying algorithm seems very sound the GUI leaves a little to be desired. I found I ended up using the commandline option more often than the GUI. Everytime you start up the GUI it loads a demo file and runs the clustering, useful if you are a first-time user who just wants to explore, but very irritating in the longer term. When you use a new input set the clustering is automatically run using the default values, if you want to alter the options you then have to rerun it.
Exploring the resulting dendrogram is very easy simply double click on a node and all the descendants of the node are displayed, whilst it is relatively easy to pick out the substructure it might have been nice to have it colour coded. In addition to the structure numeric data is also displayed.
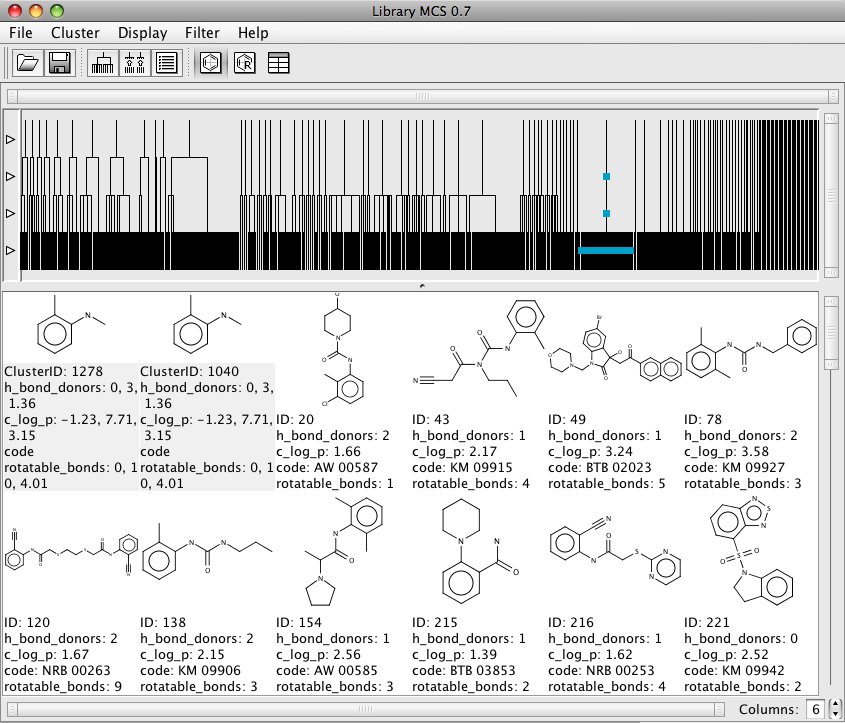
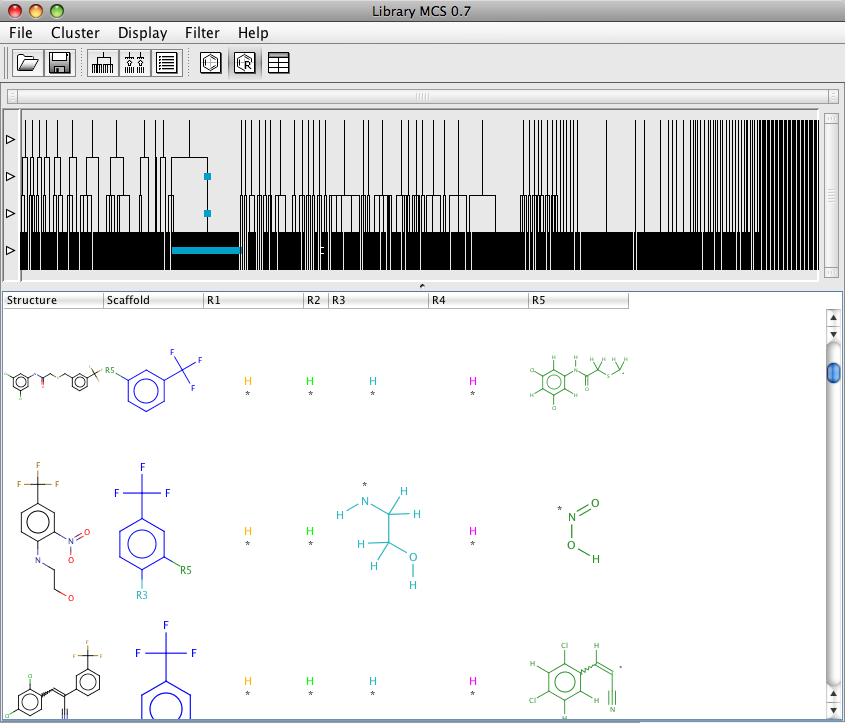
It is also possible to display the results in a table view that gives an easy way to browse through the results and the associated numeric data. However sorting the table does not seem that reliable. There is also a R-group display which might be useful for exploring SAR, however currently this view does not display the numeric data. It also does not appear to be possible to save the resulting graph.
It is difficult to give accurate benchmarks, actual speed highly depends on kind and molecular size of compounds. Diverse sets cluster much slower than more focused ones, however I ran a number datasets of several thousand structures on my laptop very easily. But remember by default, the heap size in some Java runtime environments is limited to 64MB, so you may run out of memory and need to increase the heap size. In summary, libmcs seems to be an excellent algorithm for generating MCS based clustering, the GUI however still needs some work.
LibraryMCS is part of JKluster, a Chemaxon module for diversity calculation and clustering integrated into JChem. Although the users of JChem are mainly chemists, JKlustor can be used for other objects too.
Currently JKlustor offers the following command-line tools for clustering:
- GenerateMD generates various molecular descriptors including chemical hashed and pharmacophore fingerprints for molecules, which may be used for structural diversity computations and clustering.
- Jarp performs variable-length Jarvis-Patrick clustering.
- Ward clusters molecules using Ward’s hierarchic clustering method applying the RNN approach.
- LibMCS clusters molecules by maximum common substructures in a hierarchical manner. It can be applied to focused set profiling and diversity analysis.
- CreateView composes an SDfile that contains both structures and calculation results using the input SDfile of GenerateMD and a table containing the ordinal number of compounds from the SDfile and other data to be viewed. Such table can be created for example by Compr, Jarp or Ward. The generated SDfiles can be displayed by MarvinView or other SDF viewer.
Last updated 13 March 2023