I first became aware of ChemDoodle several years ago when it was developed as an inexpensive chemical drawing package, over the years it has been refined and new features added culminating with the recent release of version 5. It is now used in 1000’s of institutions throughout the world and the beautifully detailed chemical drawings are available on Mac, Linux and Windows platforms. ChemDoodle also contains an entire interface for creating ChemDoodle Web Components. The ChemDoodle Web Components library is an open-source and free HTML5 toolkit. These components can be images, animations or interactive components and will help you create powerful content for desktop and mobile browsers. This means ChemDoodle is the ideal tool for providing chemical informations across multiple desktop and mobile platforms without the need for plugins or applets.
My first impression when starting up ChemDoodle 5 was that everything seemed very familiar but on second glance it is clear that many of the icons have been enhanced, also after a few mins clicking buttons my impression is that everything responds a little snappier. ChemDoodle is a pretty intuitive application to use but I note the User Guide has now expanded to 215 pages, and there is also a very useful 5 page summary of keyboard shortcuts. The keyboard shortcuts are a way to rapidly modify and build structures and when mastered greatly improve productivity. The ChemDoodle website also has a series of tutorial videos.
One feature that I’m sure will be enormously popular is file chooser preview. As shown below when you choose to open a file, there is a preview of the file contents shown in the small window below the file listing. This gives you a way to look at the files without the need to open the file.
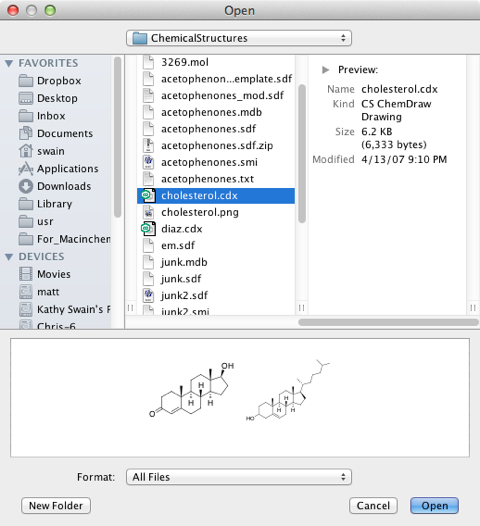
I tested this with a variety of different file types including ChemDoodle native format (.icl), CambridgeSoft (.cdx) as well as SMILES (.smi), MDL (.sdf and .mol) and Tripos (.mol2) for both 2D and 3D representations, and all displayed fine in the preview window. Files containing multiple structures will display all structures up to a file size limit (200K). If you try to preview pdb files it does seem to give an unexpected results however, each of the amino acids displays as a disconnected fragment, however the file opens correctly. Apparently a fix for these cosmetic bugs is in the works.
ChemDoodle supports a wide variety of chemical file types including
1. ChemDoodle Documents (.icl)
2. ChemDoodle Javascript Data (.cwc.js)
3. ACD/ChemSketch Document (.sk2)
4. Beilstein ROSDAL (.ros)
5. CambridgeSoft ChemDraw Exchange (.cdx)
6. CambridgeSoft ChemDraw XML (.cdxml)
7. CHARMM CARD File (.crd)
8. ChemAxon Marvin Document (.mrv)
9. Chemical Markup Language (.cml)
10. Daylight SMILES (.smi, .smiles)
11. IUPAC InChI (.inchi)
12. IUPAC JCAMP-DX (.jdx, .dx)
13. ISIS Sketch File (.skc)
14. ISIS Sketch Transportable Graphics File (.tgf)
15. MDL MOLFiles, both V2000 and V3000 connection tables (.mol, .mdl)
16. MDL RXNFiles, both V2000 and V3000 connection tables (.rxn)
17. MDL RDFiles (.rdf, .rd)
18. MDL SDFiles (.sdf, .sd )
19. Molinspiration JME String (.jme)
20. RCSB Protein Data Bank Files (.pdb, .ent)
21. Schrödinger MacroModel (.mmd, .mmod)
22. Schrödinger Maestro (.mae)
23. Standard Molecular Data (.smd)
24. Tripos Mol2 (.mol2, .ml2, .sy2)
25. Tripos Sybyl Line Notation (.sln)
26. XYZ Files (.xyz)
However some file types may contain none chemical information that is not always retained.
Widgets
By default widgets now sit in a “widget dock” at the bottom right of the main display, If you hover over any of the icons, that icon will be enlarged, and the title of the widget will appear. As well as acting as the button to activate a widget you can drag a structure onto a widget to activate it. For example dragging a structure onto the NMR SignalSeek widget will cause it to open displaying the spectra of the dragged structure.
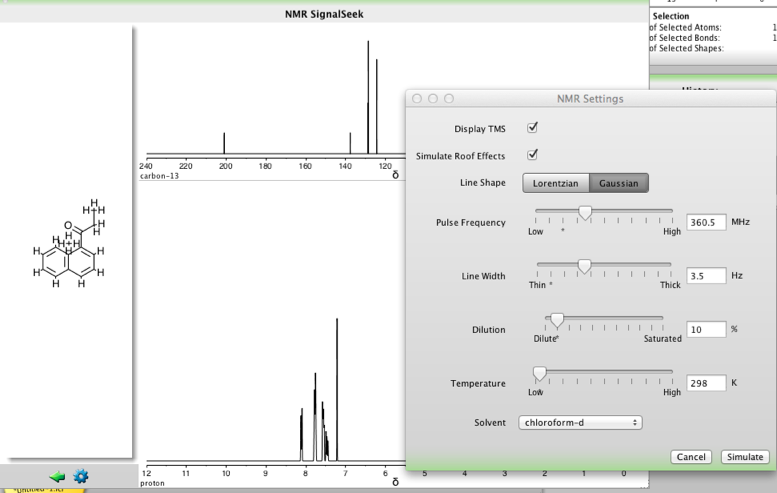
NMR SignalSeek widget simulates both 13C and 1H NMR spectra, one feature I found particularly useful was the ability to change the settings as shown in the dialog below. Peaks and nuclei are hot linked. Hovering over a peak will highlight the corresponding nuclei and vice versa. The chemical shift of the nuclei is displayed above the structure in the main structure panel. Full details of the spectra simulation are provided in the reference section of the user guide.
In addition to the NMR SignalSeek widget there is also a Multiplet Tool widget, a tool for generating multiplet trees, simulating signals and producing the figures.
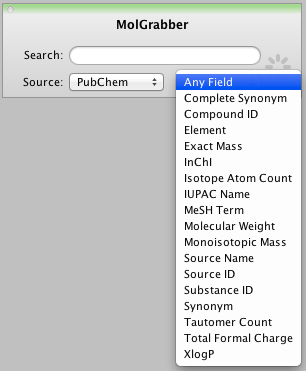
MolGrabber the widget that is used getting structures from PubChem now also supports ChemSpider and ChemExper. It would be nice to be able to modify this widget to access alternative data sources in particular internal databases, a published API would be very welcome. Depending on the datasource it is possible to refine your query by addressing specific fields, however it does not seem possible to query multiple fields at once.
The Search widget now has options to fine tune the search, defining the maximum size of files to be searched and how many structures to return. This is very useful to avoid trying to search very large files. Since the search widget can search all the file types that ChemDoodle recognises there may be cases where you don’t want to search certain file types and it might be useful to be able to define which ones should be included in the search, at present it is possible to exclude pdb but it might be useful to exclude others.
The Properties widget calculates a number of properties for a molecule:- Molecular Formula, Counts of Atoms, Bonds, Rings (Frèrejacque Number, All Rings), HBD, HBA, Degree of Unsaturation, Lipinski’s Rule of 5 Violations and Wiener Index. In addition Molar Refractivity. TPSA, XlogP v2.0, Mass and Elemental Composition are determined. In addition if you right-click on a selected structure a drop down menu gives access to a wide variety of options including the ability to calculate a range of over 60 descriptors currently categorised into 4 types: constitutional, topological, physicochemical and ADME. Whilst this is a great starting point it might be useful to have a published API so users can add their own calculated properties.
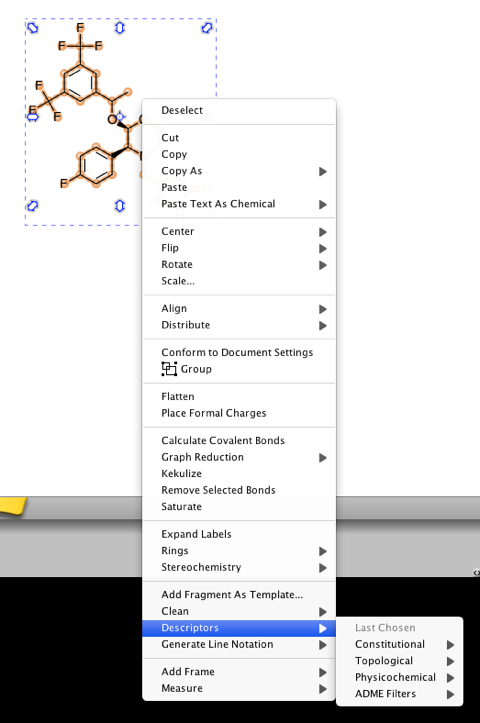
The drop down menu also allows the user to save fragments to their own Template library to build a collection of regularly used structures.
Round-trip Editing
Round-trip editing has been an issue on the Mac for a while now as applications transition from PICT to PDF format, fortunately ChemDoodle has been pretty on the ball and has supported Microsoft Word, Powerpoint and Excel for a while. Keynote is also supported but unfortunately Pages and Numbers are not supported. Interestingly if you copy a structure from Pages into Keynote and then into ChemDoodle you can edit the structure. What is happening is ChemDoodle inserts chemical information into the PDF that gets placed on the clipboard when copying a structure. The application that this PDF gets pasted into now needs to handle the PDF correctly, such that the chemical information is not stripped. Apple has this working with the latest version of Keynote. They are working towards getting the other iWorks applications to also support the expected PDF with the chemical information, so round trip editing can happen in more than Keynote. ChemDoodle also offers an alternative method to achieve round-trip editing in Pages
1. When working with figures pasted into Pages from ChemDoodle, open the Inspector by clicking on the Inspector button in Pages top toolbar.
2. In the Inspector, click on the Metrics icon , and then click on the ChemDoodle figure so that droppedImage.pdf appears.
3. Drag the droppedImage.pdf onto the Doodle Board in ChemDoodle to recover the data.
The roundtrip editing also works with FileMakerPro. Some applications may not paste the PDF by default. In this case, make sure to use a provided Paste Special function in the receiving application to choose the PDF. Not all applications on Mac OS X preserve PDF comments, and round trip editing is not possible with those applications.
Toolbars
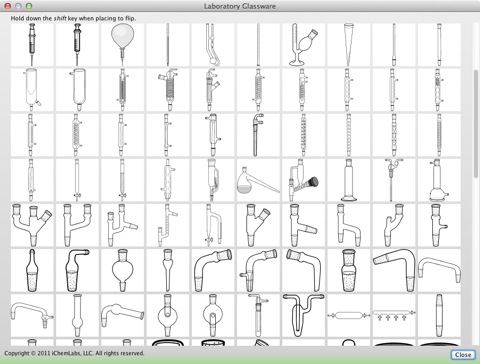
Slightly confusingly the “Shapes” toolbar contains a link to the glassware and equipment templates.
This contains a vast array of glassware and bench top equipment
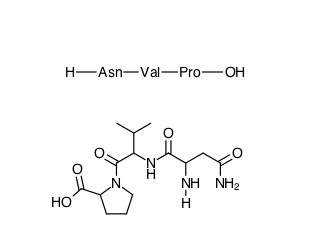
The Shapes toolbar also contains the tool for building amino acid sequences, you can build the sequence using the single letter amino-acid codes and it will be displayed as shown below. If you then right-click on the sequence and choose “Expand Labels” from the drop down menu the structure is displayed, however I couldn’t find a way to generate the correct stereochemistry.
The shape system has been updated allowing more precise control, I have to confess this is not a function I use regularly but it seems to function as one might expect.
Fragmentation Tools
One of the new features in ChemDoodle 5 is a collection of fragmentation tools in the “Content” toolbar”, there are three fragmentation functions. Molecular Weight -The molecules will be fragmented and each fragment will have a molecular weight assigned. Synthesis The molecules will be fragmented and each fragment will be set as a reactant in a synthetic reaction leading to the original molecules. Retrosynthesis The molecules will be fragmented and each fragment will be set as a product in the retrosynthetic reaction resulting in the original molecules.

Summary
This is a very good upgrade that focuses on improving workflow productivity as well as adding several useful new features, I’m sure the file preview tool will rapidly become indispensable.
There are a variety of licensing and upgrade pricing options are available, so it is worth checking the website to work out the best option for you.
Last Updated 5 November 2012