AlphaFold2 is an artificial intelligence (AI) program developed by Alphabets’s/Google’s DeepMind which performs predictions of protein structure. Despite the name AlphaFold2 does not actually predict the folding mechanism instead it predicts the final 3D structure of a protein from the protein sequence DOI.
Source code for the AlphaFold model, trained weights and inference script are available under an open-source license at https://github.com/deepmind/alphafold.
It is possible to get easy access to AlphaFold2 via a Google Colab notebook here https://colab.research.google.com/github/sokrypton/ColabFold/blob/main/AlphaFold2.ipynb however there is a 2 hour timeout, and in my testing a many of the runs timed out.
Fortunately there it is possible to run the notebook locally on your machine, as written in a brilliant description by Yoshitaka Moriwaki https://github.com/YoshitakaMo/localcolabfold.
Installing LocalColabfold
There are instructions for multiple platforms but I thought I’d show details and pictures for installing on Apple Silicon, I’m using a MacBook Pro M1 max with 64GB memory under macOS 12.1
Firstly install Home-brew if not already installed. (Homebrew is a free and open-source software package management system that simplifies the installation of software on Macs).
/bin/bash -c "$(curl -fsSL https://raw.githubusercontent.com/Homebrew/install/HEAD/install.sh)"
Then install a couple of packages
brew install wget cmake gnu-sed
brew install brewsci/bio/hh-suite
The next step is to create a folder called Alphafold then in the Terminal type
cd /Users/chrisswain/Projects/Alphafold
To enter the newly created folder and then install miniconda using Home-brew
brew install --cask miniforge
Then download the colabfold download/install script
wget https://raw.githubusercontent.com/YoshitakaMo/localcolabfold/main/install_colabbatch_M1mac.sh
You should now have a file called installcolabbatchM1mac.sh
bash install_colabbatch_M1mac.sh
After a few minutes a new folder should have been created as shown below.
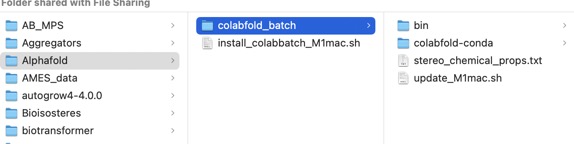
When I tried to run the program I got an error saying SciPy was not installed, so I installed it using the colabfold conda
colabfold-conda/bin/python3.8 -m pip install scipy --no-deps --no-color
This has been corrected in the latest commit https://github.com/YoshitakaMo/localcolabfold/issues/55
If you now view the help by typing the command
./bin/colabfold_batch -h
/Users/chrisswain/Projects/Alphafold/colabfold_batch/colabfold-conda/lib/python3.8/site-packages/jax/_src/lib/__init__.py:32: UserWarning: JAX on Mac ARM machines is experimental and minimally tested. Please see https://github.com/google/jax/issues/5501 in the event of problems.
warnings.warn("JAX on Mac ARM machines is experimental and minimally tested. "
usage: colabfold_batch [-h] [--stop-at-score STOP_AT_SCORE] [--num-recycle NUM_RECYCLE] [--num-models {1,2,3,4,5}] [--recompile-padding RECOMPILE_PADDING]
[--model-order MODEL_ORDER] [--host-url HOST_URL] [--data DATA] [--msa-mode {MMseqs2 UniRef+Environmental),MMseqs2 (UniRef only,single_sequence}]
[--model-type {auto,AlphaFold2-ptm,AlphaFold2-multimer}] [--amber] [--templates] [--env] [--cpu] [--rank {auto,plddt,ptmscore,multimer}]
[--pair-mode {unpaired,paired,unpaired+paired}] [--recompile-all-models] [--sort-queries-by {none,length,random}] [--zip] [--overwrite-existing-results]
input results
positional arguments:
input Can be one of the following: Directory with fasta/a3m files, a csv/tsv file, a fasta file or an a3m file
results Directory to write the results to
optional arguments:
-h, --help show this help message and exit
--stop-at-score STOP_AT_SCORE
Compute models until plddt or ptmscore > threshold is reached. This can make colabfold much faster by only running the first model for easy queries.
--num-recycle NUM_RECYCLE
Number of prediction cycles.Increasing recycles can improve the quality but slows down the prediction.
--num-models {1,2,3,4,5}
--recompile-padding RECOMPILE_PADDING
Whenever the input length changes, the model needs to be recompiled, which is slow. We pad sequences by this factor, so we can e.g. compute sequence from length 100 to 110 without recompiling. The prediction will become marginally slower for the longer input, but overall performance increases due to not recompiling. Set to 1 to disable.
--model-order MODEL_ORDER
--host-url HOST_URL
--data DATA
--msa-mode {MMseqs2 (UniRef+Environmental),MMseqs2 (UniRef only),single_sequence}
Using an a3m file as input overwrites this option
--model-type {auto,AlphaFold2-ptm,AlphaFold2-multimer}
predict strucutre/complex using the following model.Auto will pick "AlphaFold2" (ptm) for structure predictions and "AlphaFold2-multimer" for complexes.
--amber Use amber for structure refinement
--templates Use templates from pdb
--env
--cpu Allow running on the cpu, which is very slow
--rank {auto,plddt,ptmscore,multimer}
rank models by auto, plddt or ptmscore
--pair-mode {unpaired,paired,unpaired+paired}
rank models by auto, unpaired, paired, unpaired+paired
--recompile-all-models
recompile all models instead of just model 1 ane 3
--sort-queries-by {none,length,random}
sort queries by: none, length, random
--zip zip all results into one <jobname>.result.zip and delete the original files
--overwrite-existing-results
To generate a 3D protein structure you need a protein sequence in fasta format
These can be obtained from the Uniprot database, for example HUMAN Free fatty acid receptor 2 https://www.uniprot.org/uniprot/O15552
>sp|O15552|FFAR2_HUMAN Free fatty acid receptor 2 OS=Homo sapiens OX=9606 GN=FFAR2 PE=1 SV=1 MLPDWKSSLILMAYIIIFLTGLPANLLALRAFVGRIRQPQPAPVHILLLSLTLADLLLLL LLPFKIIEAASNFRWYLPKVVCALTSFGFYSSIYCSTWLLAGISIERYLGVAFPVQYKLS RRPLYGVIAALVAWVMSFGHCTIVIIVQYLNTTEQVRSGNEITCYENFTDNQLDVVLPVR LELCLVLFFIPMAVTIFCYWRFVWIMLSQPLVGAQRRRRAVGLAVVTLLNFLVCFGPYNV SHLVGYHQRKSPWWRSIAVVFSSLNASLDPLLFYFSSSVVRRAFGRGLQVLRNQGSSLLG RRGKDTAEGTNEDRGVGQGEGMPSSDFTTE
Save the file as ffa2.fasta
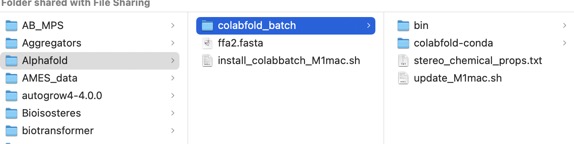
We can now run a prediction thus
./bin/colabfold_batch --amber --templates --num-recycle 3 --cpu /Users/chrisswain/Projects/Alphafold/ffa2.fasta FFA2output
You will get warnings about this being minimally tested on ARM machines
On your first run AlphaFold2 weight parameters will be downloaded at ~/Library/Caches/colabfold/params directory in subsequent runs these will not be downloaded again.
/Users/chrisswain/Projects/Alphafold/colabfold_batch/colabfold-conda/lib/python3.8/site-packages/jax/_src/lib/__init__.py:32: UserWarning: JAX on Mac ARM machines is experimental and minimally tested. Please see https://github.com/google/jax/issues/5501 in the event of problems.
warnings.warn("JAX on Mac ARM machines is experimental and minimally tested. "
WARNING: You are welcome to use the default MSA server, however keep in mind that it's a limited shared resource only capable of processing a few thousand MSAs per day. Please submit jobs only from a single IP address. We reserve the right to limit access to the server case-by-case when usage exceeds fair use.
If you require more MSAs, please host your own API and pass it to --host-url
2022-02-12 19:41:35,703 Running colabfold 1.2.0 (ae2b519f4483253dc2790c1545ce94b922eaa07b)
2022-02-12 19:41:35,717 Found 8 citations for tools or databases
2022-02-12 19:41:39,408 Query 1/1: sp_O15552_FFAR2_HUMAN_Free_fatty_acid_receptor_2_OS_Homo_sapiens_OX_9606_GN_FFAR2_PE_1_SV_1 (length 330)
COMPLETE: 100%| ███████████████████████████████████████████████████████████████████████████████████████████████████████████████████████| 150/150 [elapsed: 00:00 remaining: 00:00]
2022-02-12 19:41:53,187 Sequence 0 found templates: [b'6ibb_A' b'2ksb_A' b'6c1q_B' b'6osa_R' b'4n6h_A' b'5w0p_C' b'6wwz_R'
b'5w0p_D' b'6cmo_R' b'6lfm_R' b'6lfo_R' b'2z73_A' b'3ayn_B' b'5dhh_B'
b'6c1r_B' b'6ko5_A' b'5dhg_B' b'6b73_A' b'5yhl_A' b'5ywy_A']
2022-02-12 19:41:53,411 Running model_3
2022-02-12 19:41:54.516147: W tensorflow/core/platform/profile_utils/cpu_utils.cc:128] Failed to get CPU frequency: 0 Hz
2022-02-12 20:10:34,625 model_3 took 1719.6s (3 recycles) with pLDDT 83.8
/Users/chrisswain/Projects/Alphafold/colabfold_batch/colabfold-conda/lib/python3.8/site-packages/simtk/__init__.py:2: UserWarning:
You are using an experimental build of OpenMM v7.5.1.
This is NOT SUITABLE for production!
It has not been properly tested on this platform and we cannot guarantee it provides accurate results.
warnings.warn("""
2022-02-12 20:10:51,826 Running model_4
2022-02-12 20:37:14,646 model_4 took 1581.9s (3 recycles) with pLDDT 78
2022-02-12 20:37:28,925 Running model_5
2022-02-12 21:03:45,912 model_5 took 1575.7s (3 recycles) with pLDDT 82.5
2022-02-12 21:04:00,337 Running model_1
2022-02-12 21:06:55.622754: E external/org_tensorflow/tensorflow/compiler/xla/service/slow_operation_alarm.cc:55]
********************************
Very slow compile? If you want to file a bug, run with envvar XLA_FLAGS=--xla_dump_to=/tmp/foo and attach the results.
Compiling module jit_apply_fn__1.134522
*******************************
2022-02-12 21:34:16,130 model_1 took 1814.7s (3 recycles) with pLDDT 83.9
2022-02-12 21:34:31,196 Running model_2
2022-02-12 22:02:11,364 model_2 took 1659.1s (3 recycles) with pLDDT 79.7
2022-02-12 22:02:26,548 reranking models by plddt
2022-02-12 22:02:27,280 Done
You should now have an output like this.
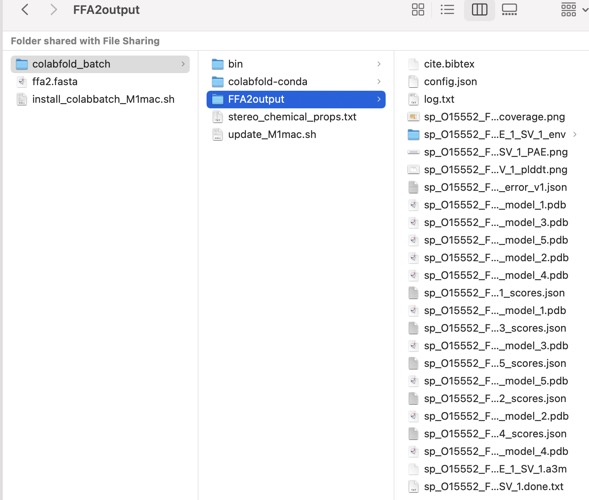
This folder contains various files, the “env” folder contains the templates used. The log file containing the timings from each of the models There are also the unrelaxed PDB files of the direct output from the models, a PDB format text file containing the predicted structure after performing an Amber relaxation procedure on the unrelaxed structure prediction. Plus the images shown below.
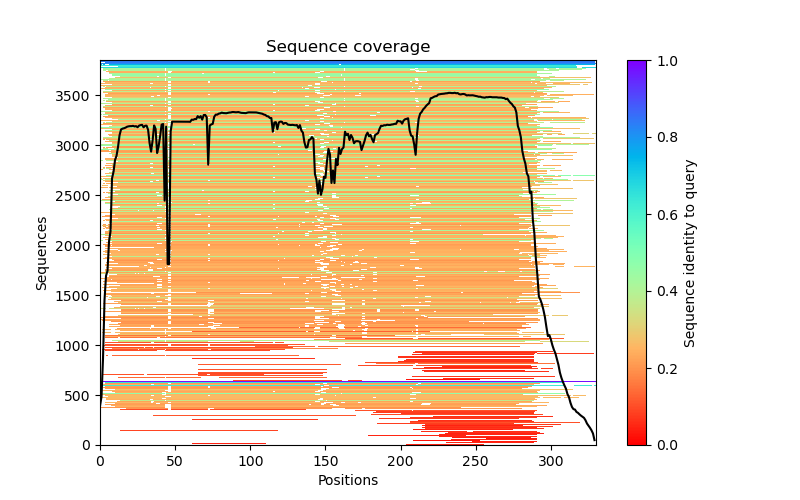
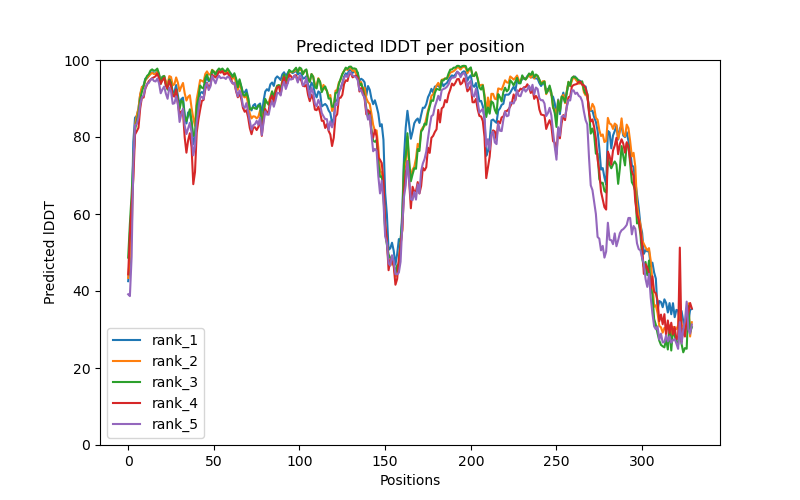
f you open the PDB files in a viewer like ChimeraX you can display the structure as shown below. The pLDDT confidence measure is stored in the B-factor field of the output PDB files so you can colour by b-factor in ChimeraX to get a visual representation (red is high confidence, blue is low confidence).
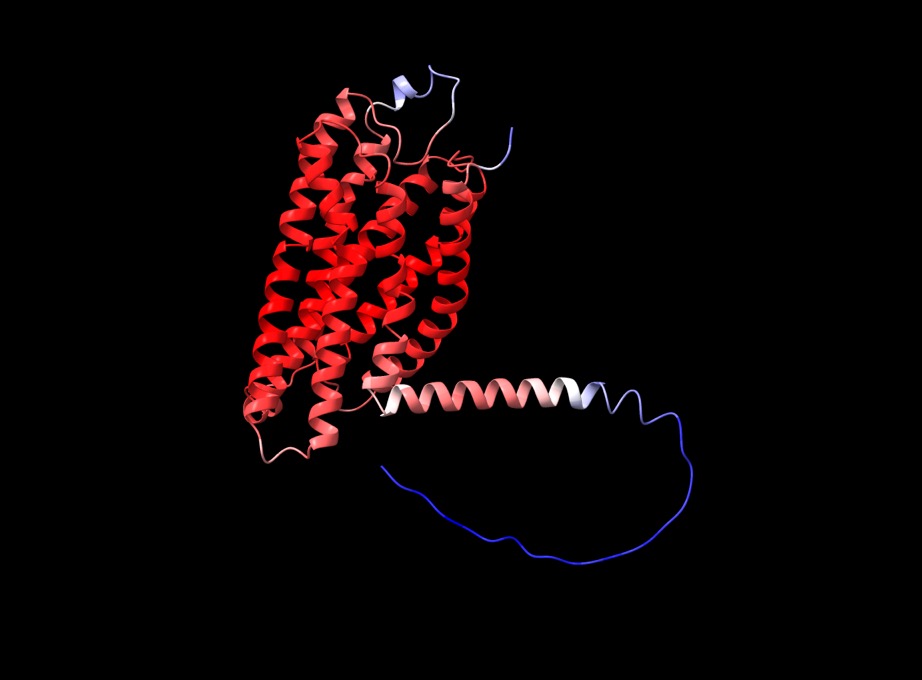
I got a couple of tips from Yoshitaka
I recommend to adding
--model-order 1,2,3,4,5argument to reduce the calculation time when one uses--templates. By Default, two JAX compilations are required when starting the calculation for model 3 and model 1.
And
Preparing the input file for complex prediction is a bit more complicated and differs from that of the original AlphaFold. Here is an example for localcolabfold:
>3kud_complex MTEYKLVVVGAGGVGKSALTIQLIQNHFVDEYDPTIEDSYRKQVVIDGETCLLDILDTAGQEEYSAMRDQYMRTGEGFLC VFAINNTKSFEDIHQYREQIKRVKDSDDVPMVLVGNKCDLAARTVESRQAQDLARSYGIPYIETSAKTRQGVEDAFYTLV REIRQH: PSKTSNTIRVFLPNKQRTVVNVRNGMSLHDCLMKALKVRGLQPECCAVFRLLHEHKGKKARLDWNTDAASLIGEELQVDF L
This input fasta file (3kud_complex.fasta) will produce complex structures.
List of tools tested https://macinchem.co.uk/software-reviews/cheminformatics-and-compchem-on-apple-silicon/
Last Updated 14 Feb 2022