A recent paper described Boltz-1: Democratizing Biomolecular Interaction Modeling https://www.biorxiv.org/content/10.1101/2024.11.19.624167v2
Boltz-1, an open-source deep learning model incorporating innovations in model architecture, speed optimization, and data processing achieving AlphaFold3-level accuracy in predicting the 3D structures of biomolecular complexes. Boltz-1 demonstrates a performance on-par with state-of-the-art commercial models on a range of diverse benchmarks, setting a new benchmark for commercially accessible tools in structural biology.
I tried installing Boltz-1 on Apple Silicon and whilst I did get it running the it was far from straight-forward. Fortunately, the current version of ChimeraX 1.10 will install Boltz-1 on a Mac. Making Boltz Structure Predictions in ChimeraX https://www.rbvi.ucsf.edu/chimerax/data/boltz-apr2025/boltz_help.html A ChimeraX graphical user interface (menu Tools / Structure Prediction / Boltz) and ChimeraX command (boltz) are provided to make predictions.
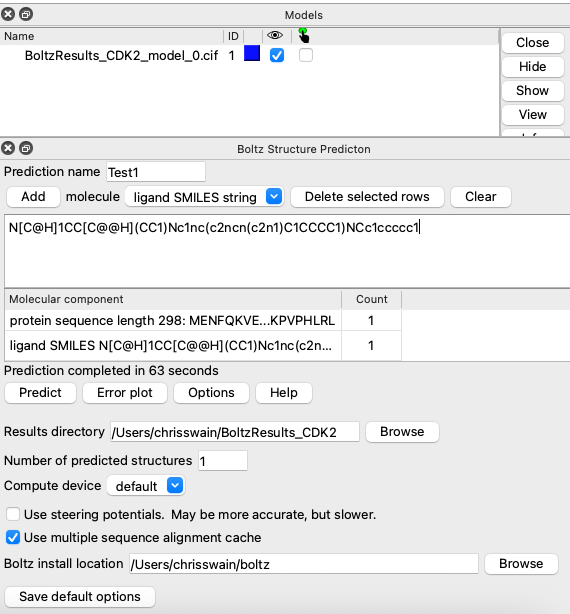
Whilst this is an excellent way to run the docking I thought it might be interesting to look at accessing the process from other applications, in particular chemical drawing applications like ChemDraw. Looking at the process that is used by ChimeraX to run Boltz there are a number of key features
The application Boltz as installed by ChimeraX is in the user home directory in my case this is /Users/chrisswain/boltz/bin/boltz
The input data is stored in a yaml file.
After running a prediction from within ChimeraX the cached multiple sequence alignments in my case are in the downloads folder by default.
To run a prediction from ChemDraw we need to create the yaml file for which we need the protein sequence, the location of the msa, and the SMILES string of the proposed ligand.
The first part of the script creates a folder in the users home directory to write the yaml file to. The next part uses ChemDraw to generate the SMILES string from the drawn structure, we then get a name for the run. The next step generates the text for the yaml file (you will need to edit the path to the msa file), this is then written to the folder created in the first step. We then create the input command and then run it through the Terminal. The results will be in a folder called “boltz_results_Run1” (or whatever name you have chosen for the run).
I stored the script in the Script Menu (https://support.apple.com/en-gb/guide/script-editor/scpedt27975/mac) activated from within the script editor.
The Applescript
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 |
--Created by Macs in Chemistry (http://www.macinchem.org) tell application "Finder" set targetfolder to (path to home folder as string) & "BoltzResults_CDK2" display dialog targetfolder set fileExists to exists of targetfolder display dialog fileExists if fileExists = false then make new folder at (path to home folder as string) with properties {name:"BoltzResults_CDK2"} end if end tell set the clipboard to "" --This could be modified to any application that can generate the SMILES string. tell application "ChemDraw 23.0.1" activate if not (enabled of menu item "Copy") then do menu item "Select All" of menu "Edit" end if do menu item "SMILES" of menu "Copy As" of menu "Edit" set theSMILES to the clipboard display dialog theSMILES end tell --display dialog theSMILES set theResponse to display dialog "Name for docking run" default answer "Run1" with icon note buttons {"Cancel", "Continue"} default button "Continue" set theFileName to text returned of theResponse #yml text set the yml_text to "version: 1 sequences: - protein: id: [A] sequence: MENFQKVEKIGEGTYGVVYKARNKLTGEVVALKKIRLDTETEGVPSTAIREISLLKELNHPNIVKLLDVIHTENKLYLVFEFLHQDLKKFMDASALTGIPLPLIKSYLFQLLQGLAFCHSHRVLHRDLKPQNLLINTEGAIKLADFGLARAFGVPVRTYTHEVVTLWYRAPEILLGCKYYSTAVDIWSLGCIFAEMVTRRALFPGDSEIDQLFRIFRTLGTPDEVVWPGVTSMPDYKPSFPKWARQDFSKVVPPLDEDGRSLLSQMLHYDPNKRISAKAALAHPFFQDVTKPVPHLRL msa: /Users/chrisswain/Downloads/ChimeraX/BoltzMSA/CDK2/CDK2_0.csv - ligand: id: [B] smiles: " & theSMILES set this_file to targetfolder & ":" & theFileName & ".yaml" my write_to_file(yml_text, this_file, false) on write_to_file(this_data, target_file, append_data) try set the target_file to the target_file as text set the open_target_file to ¬ open for access file target_file with write permission if append_data is false then ¬ set eof of the open_target_file to 0 write this_data to the open_target_file starting at eof close access the open_target_file --display dialog "file_done" return true on error try close access file target_file end try return false end try end write_to_file --display dialog yml_text set boltzyaml to POSIX path of this_file set boltzScript to "/Users/chrisswain/boltz/bin/boltz predict " & boltzyaml & " --write_full_pae --accelerator gpu --no_potentials" tell application "Terminal" activate set currentTab to do script boltzScript end tell |
This should be regarded as a preliminary work in progress, I had hoped I could store the script in the Chemdraw scripts folder but I got an error saying ChemDraw did not have access to send events to another application and I’m not sure how to change that.
Rather than using a drawing package this script could also be modified to import a list of SMILES strings and then execute Boltz runs on each of them in turn.
There is an update to Boltz that may require modifications to this script and the folks at ChimeraX are already looking at implementing it. I’ll also look at improving the script.