Hydrogen bonds are critical for many aspects of chemistry and biology, from the physicochemical properties of molecules, binding affinity, membrane permeability and more. However, prediction of hydrogen bonding strength is challenging.
A recent preprint from Rowan Scientific on Chemrxiv https://chemrxiv.org/engage/chemrxiv/article-details/67916aec81d2151a0274ff60 describes a methodology that requires on modest computational cost. It uses rapid conformer generation and optimization with neural network potentials, followed by a single density-functional-theory calculation of the electrostatic potential.
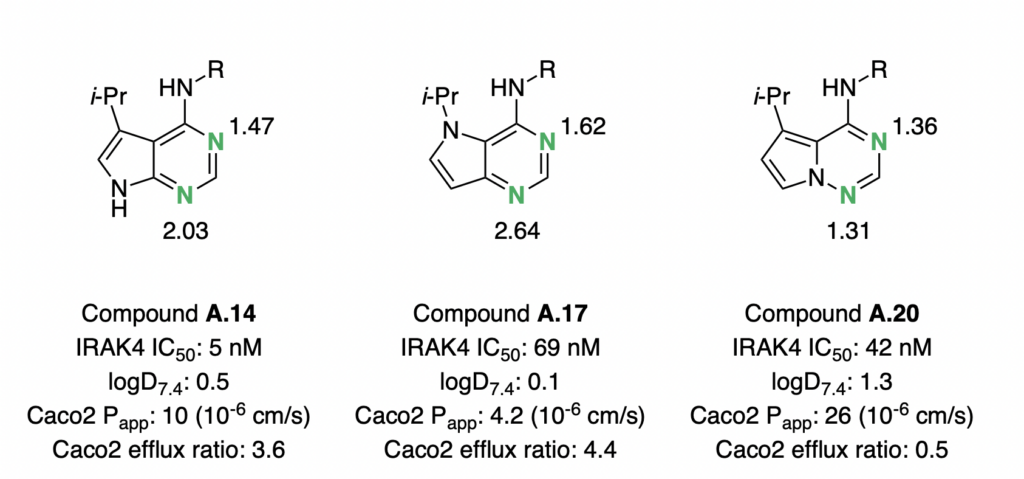
If you would like to try it out there are a number of options available https://rowansci.com/pricing including a python api.