The 2011.10 release of Chemical Computing Group’s Molecular Operating Environment (MOE) software includes a number of new features, enhancements and changes, I’ve had a quick look at the update and tried to summarise a few of the features that caught my eye.
For Mac users probably the key feature is that this update now supports Mac OS X 10.7 (Lion), strictly speaking previous versions of MOE supported Lion, it was just the license manager that did not. The 2011 release of MOE uses a different license manager than that used in previous versions of MOE. To run MOE 2011.10, $MOE/bin/lmgrd and $MOE/bin/chemcompd from the 2011 release must be used; older versions of lmgrd and chemcompd will not work. Older versions of MOE are not compatible with the new license manger but the 2011 distribution does include a copy of the 2010 executable that is compatible.
One of the features I was particularly interested in was the updated nonbonded interaction visualisation.
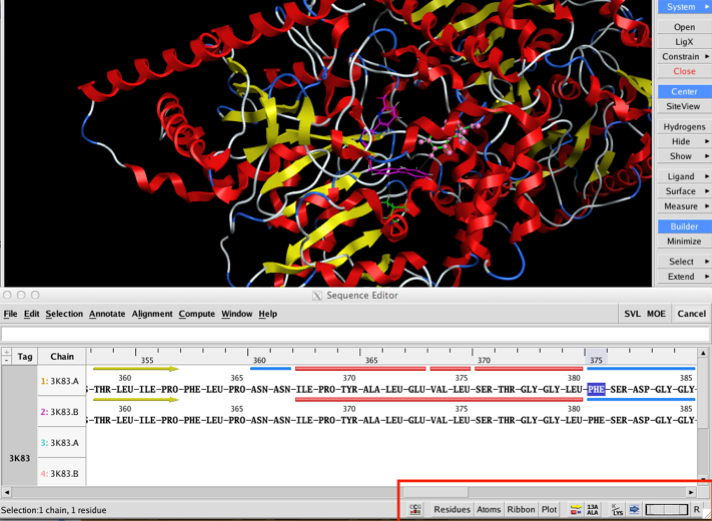
The two images below show the Crystal Structure Analysis of a Biphenyl/Oxazole/Carboxypyridine alpha-ketoheterocycle Inhibitor Bound to a Humanized Variant of Fatty Acid Amide Hydrolase (10.1021/jm9012196) 3K83. The first image shows the display using an earlier version of MOE the second shows the same complex using the latest version of MOE. As you can see arene-arene and arene-H interactions are clearly rendered, in addition the improved scoring and rendering of hydrogen bonds has highlighted extra interactions.
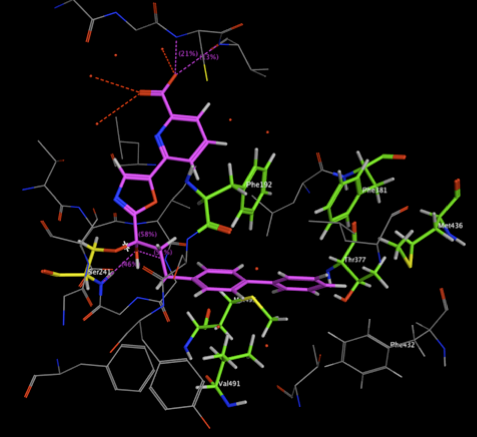
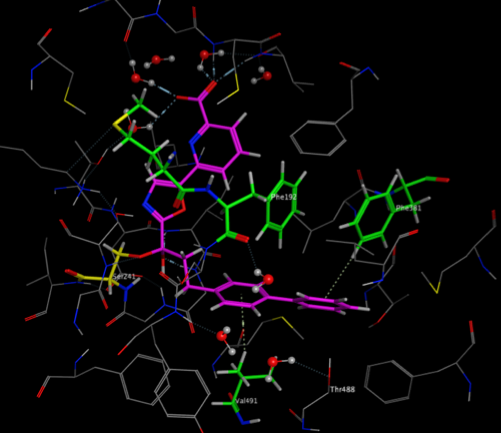
These interactions are also displayed in the 2D ligand interactions schematic view shown below.
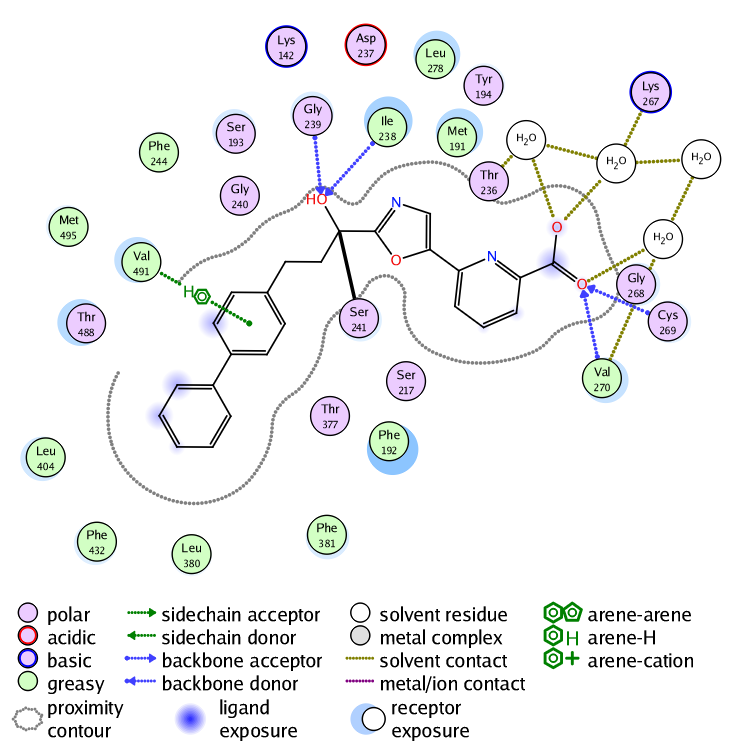
In addition a halogen bond model has been developed which agrees with crystal geometry distributions. The energy parameters from Extended Hueckel Theory are combined with an electrostatic specialised potential that is based on the “sigma hole” concept. Sulfur-LonePair interactions are supported with a model similar to that of the halogen bond. The sigma holes of divalent sulfur atoms (acting as a Lewis acid) can produce close contacts with hydrogen bond acceptors (e.g. thiophene-carbonyl) and the model is in good agreement with observed crystal structure geometries.
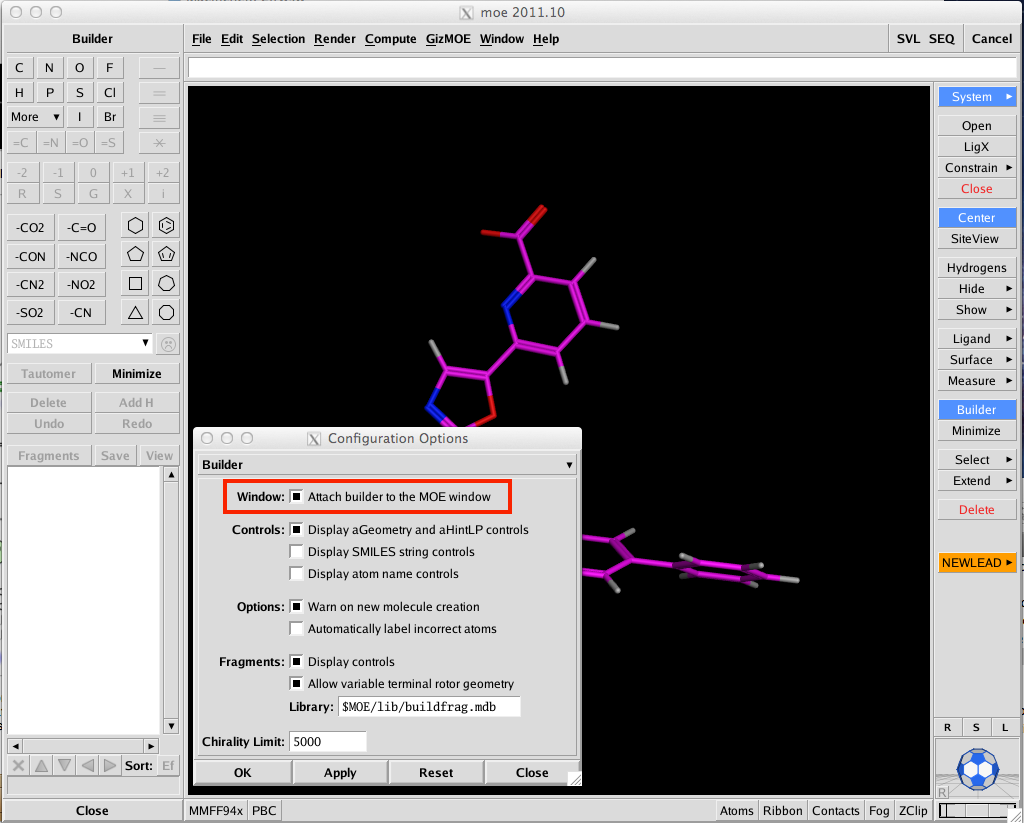
I suspect one of the most popular features will be the ability to attach the small molecule builder window to the MOE main window, if you are like me and have multiple windows open this will save time hunting around for the small builder window hidden below other windows. To activate this option simply select Window/Options from the main MOE menu, choose builder from the drop down menu and choose the appropriate checkbox. The Builder can now also scan a predefined library of R-groups — by default $MOE/lib/buildfrag.mdb — as candidate substituents for 1 or 2 selected hydrogen atoms.
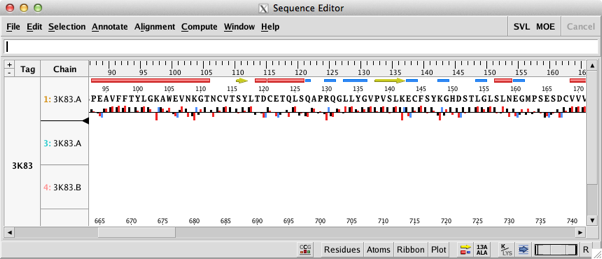
Perhaps the biggest changes are to the sequence editor; this has been completely redesigned to allow more interaction with the main display. At the bottom right of the sequence editor are a series of popup menus that allow the user to configure the display to show secondary structure, to display the residue number, change font size (I found it very useful to reduce the find size to very small to identify residues after selecting them in the main MOE window) and to change the display in the main MOE window. This is now a great way to pick out key residues in an active site. One nice feature is that when you mouse over a residue the name and sequence id appear in the footer. The main menu bar has been reorganised with new Annotate, Align and Compute menus. New functionality exists for identifying important residue segments in Kinases, Antibodies and GPCRs; automatic alignment constraints can be added for these classes as well.
The new controls allow for display of single letter residues, to hide chains and to annotate with predicted solvent accessibility.
Those interested in modelling small molecules docked into proteins will be interested to see a new forcefield has been added to MOE. It is a combination of the standard AMBER10 parameter set (parm99 + frcmod.ff99sb + fcmod.parmbsc0) with the recently released parm@frosst small molecule parameters developed over many years at Merck-Frosst by (C. Bayly, D. McKay and J-F. Truchon). These small molecule parameters are fully compatible with the Amber protein parameters. The forcefield file is $MOE/lib/amber10p.ff and is available in the Potential Setup window. The associated dictionary charges are in $MOE/lib/amber10.mdb which contains Amber standard amino acids and nucleic acids but also many common non-standard residues and cofactors. RESP charges were calculated for each of the additional residues.
The non-redundant PDB databases included with MOE have been updated and now includes over 28,000 entries derived from all entries in the PDB up to September 26, 2011, in addition the antibody and kinase databases previously released now include a GPCR database. The GPCR database contains 157 entries at the moment with associated ligands etc. All structures in the database have been superimposed in the backbone atoms of the TM-helix region to the corresponding region of a reference structure thus creating a unified frame of reference . This assists in elucidation of subtle structural differences within the Class A receptors.
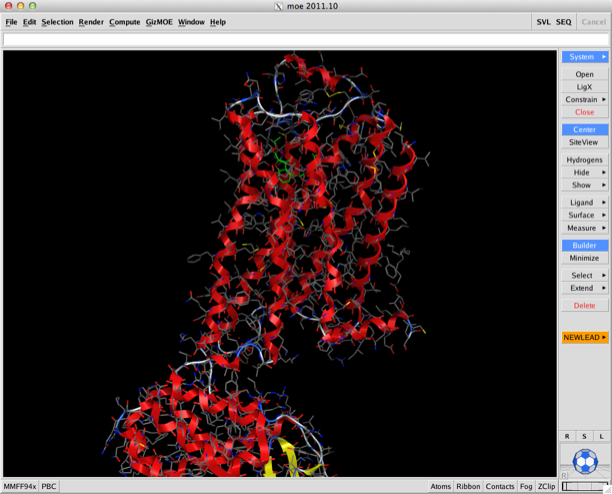
Whilst I’ve not had a chance to look at it the addition of the ability to do an analysis of solvent in binding sounds intriguing. This allows you to calculate within minutes a solvent binding free energy map using 3D-RISM, calculate water, salt and hydrophobe solvation densities in complex or apo receptor, diagnose how well alternate groups take advantage of water upon binding.
All in all this a solid update with many useful additions particularly for those involved in ligand design.
Updated 30 December 2011