I’ve recently become interested the comparison of the amino amino-acid composition of peptides, to allow comparison of cyclic versus linear peptides, or brain penetrant verses non-penetrant. I had a look around but could not find any tools that did this, in particular I wanted to include any non-proteinergic amino-acids. This would include natural amino acids that are not normally incorporated into peptides but also the many synthetic amino acids that have been published in the literature.
Compiling a list of Amino-Acids
Whilst sites like SwissSidechain have a database of several hundred amino-acid structures for download a quick inspection suggests it lacks most of the synthetic amino-acids that have been published. Fortunately with the advent of HELM notation ChEMBL have compiled a list of monomers generated by fragmenting all ChEMBL peptides that contain at least three amino acids.
For the most common unnatural amino acids, we’ve used peptide vendor catalogs to derive an ID and name. Additionally, in most cases where those amino acids are capped and/or substituted at the side-chain, the monomer ID has been prefixed/suffixed with the cap name and/or extended with the information about the side-chain substitution in parentheses. As an example, the monomer ‘methyl 4-Chloro-L-phenylalanine’ can be identified by the monomer ID ‘Me_Phe(4-Cl)’.
The file can be downloaded from the ftp site ftp://ftp.ebi.ac.uk/pub/databases/chembl/ChEMBLdb/latest/file is: chembl23monomer_library.xml and it contains nearly 3000 amino-acids.
This file is in XML format as shown below
<?xml version="1.0" encoding="UTF-8"?>
<MonomerDB xmlns="lmr" xmlns:xsi="http://www.w3.org/2001/XMLSchema-instance">
<PolymerList>
<Polymer polymerType="PEPTIDE">
<Monomer>
<MonomerID>X2484</MonomerID>
<MonomerSmiles>CC(=O)SCCCC[C@H](N[*])C([*])=O |$;;;;;;;;;;_R1;;_R2;$|</MonomerSmiles>
<MonomerMolFile>H4sIAAAAAAAAAI2TTU7DMBCF9z7FSHTLaMb/swbUFQW1iAtUCPX+F2BsN7ZTUYgVRe/Fn1/G48QAnM6Xj6/vy5mYLQcmS/bZGGAP7ACoXlzvY4gIfFoiMsUxOpVNSWiKkMqzJ2gRNxxh+oMjtJFiUxzITdzbzFmMtEoZ6jRzDmXTewPae3krLmLYlJcwlUb9mydInbuTp5tdxFgTc+uCDZSnNYc5OyOFhcte/MQdHwbHhG5LrYN75NturfIYvZPryQWR9PsZct0XN6PKtX024/uMqtCNqtiNqtSNqtyNLCZWw7TMqGKeMO4VqCqf/DD+Ws4rwHH/3mplqEQBwZaZl4P+Ljsd5gd2o4VdTQMAAA==</MonomerMolFile>
<MonomerType>Backbone</MonomerType>
<PolymerType>PEPTIDE</PolymerType>
<NaturalAnalog>X</NaturalAnalog>
<MonomerName>X2484</MonomerName>
<Attachments>
<Attachment>
<AttachmentID>R1-H</AttachmentID>
<AttachmentLabel>R1</AttachmentLabel>
<CapGroupName>H</CapGroupName>
<CapGroupSmiles>[*][H] |$_R1;$|</CapGroupSmiles>
</Attachment>
<Attachment>
<AttachmentID>R2-OH</AttachmentID>
<AttachmentLabel>R2</AttachmentLabel>
<CapGroupName>OH</CapGroupName>
<CapGroupSmiles>O[*] |$;_R2$|</CapGroupSmiles>
</Attachment>
</Attachments>
</Monomer>
I needed the SMILES string and the ID and I did try to open the XML file in a couple of applications to no avail so instead I created a very basic Jupyter Notebook. Once we have defined the “root” we can use it to navigate to an element in the tree, to get the element containing the SMILES
print(root[0][0][0][1].text)
CC(=O)SCCCC[C@H](N[*])C([*])=O |$;;;;;;;;;;_R1;;_R2;$|
We can isolate just the SMILES string using
mySMILES, rest = theSMILES.split(" |")
mySMILES
'CC(=O)SCCCC[C@H](N[*])C([*])=O'

One issue is that a substructure search using the SMILES string for Alanine would also flag other amino acids that contain the alanine substructure such as Leucine or Lysine as highlighted below.
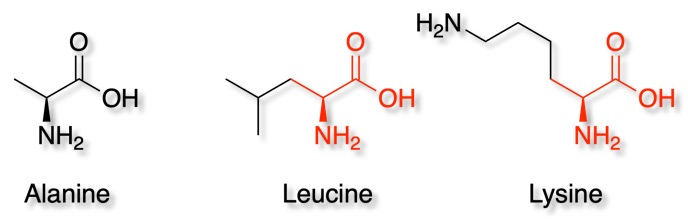
The simplest way to avoid this is to add explicit hydrogens using RDKit. First we convert the SMILES string to an RDKit molecular object, then add hydrogens, then convert back to SMILES.
m = Chem.MolFromSmiles(mySMILES)
m2=Chem.AddHs(m)
ExplicitSMILES = Chem.MolToSmiles(m2)
ExplicitSMILES
'*C(=O)[C@@]([H])(N(*)[H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])SC(=O)C([H])([H])[H]'
The result is shown below
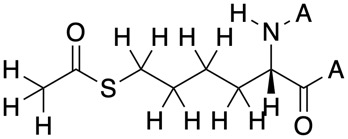
We can now loop through the XML file, extracting the SMILES and the ID, adding explicit hydrogens to the SMILES string and then creating a list of all Explicit SMILES and associated ID. This list can then be exported to a file.
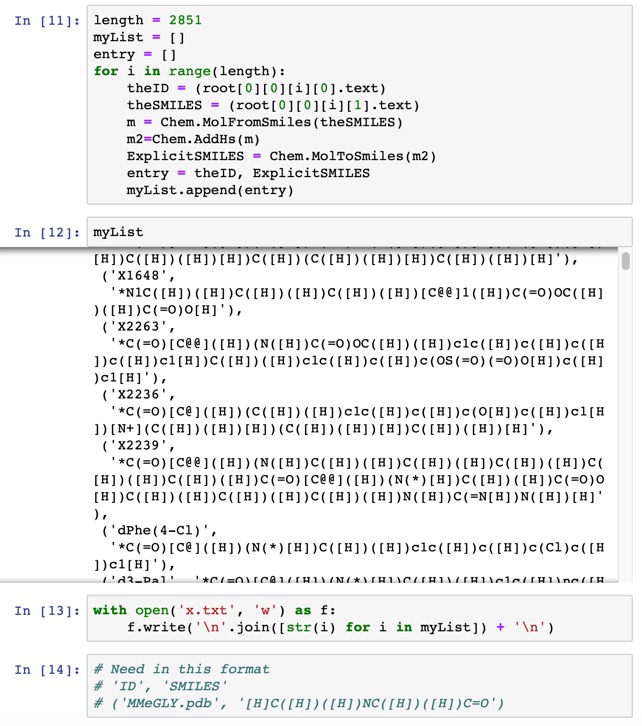
The file was then converted to the format needed Vortex by editing in BBEdit and then saved as SMARTS.txt
('X2484', '*C(=O)[C@@]([H])(N(*)[H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])SC(=O)C([H])([H])[H]'),
('X414', '*C(=O)C([H])([H])N(*)c1c([H])c([H])c([H])c([H])c1[H]'),
('X508', '*C(=O)[C@]([H])(N(*)[H])C([H])([H])C([H])([H])C(F)(F)F'),
('Aib_OMe', '*N([H])C(C(=O)OC([H])([H])[H])(C([H])([H])[H])C([H])([H])[H]'),
('Boc_V', '[H]N(C(=O)OC(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H])[C@]([H])(C(*)=O)C([H])(C([H])([H])[H])C([H])([H])[H]'),
('X1648', '*N1C([H])([H])C([H])([H])C([H])([H])[C@@]1([H])C(=O)OC([H])([H])C(=O)O[H]'),
('X2263', '*C(=O)[C@@]([H])(N([H])C(=O)OC([H])([H])c1c([H])c([H])c([H])c([H])c1[H])C([H])([H])c1c([H])c([H])c(OS(=O)(=O)O[H])c([H])c1[H]'),
('X2236', '*C(=O)[C@]([H])(C([H])([H])c1c([H])c([H])c(O[H])c([H])c1[H])[N+](C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H]'),
('X2239', '*C(=O)[C@@]([H])(N([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C(=O)[C@@]([H])(N(*)[H])C([H])([H])C(=O)O[H])C([H])([H])C([H])([H])C([H])([H])N([H])C(=N[H])N([H])[H]'),
('dPhe(4-Cl)', '*C(=O)[C@]([H])(N(*)[H])C([H])([H])c1c([H])c([H])c(Cl)c([H])c1[H]'),
('d3-Pal', '*C(=O)[C@]([H])(N(*)[H])C([H])([H])c1c([H])nc([H])c([H])c1[H]'),
Substructure searching in Vortex
In the tutorial Scripting Vortex 37 the script flags the presence (or absence) of a variety of functional groups by matching SMARTS strings to provide categorisation of potential reagents/starting materials for reaction workflows. In this script we will use the same strategy using the amino-acid SMILES as queries and writing a flag for the presence (or absence) for each of the amino-acids for all the peptides in the workspace.
The first part of the script sets up the search to use multiple processors, we then read in the SMARTS patterns form the SMARTS.txt file. The script then generates the SMILES strings for the peptides in the workspace if none is present. It then runs multiple SMARTS matching in parallel, creating a new column in the workspace for each amino-acid.
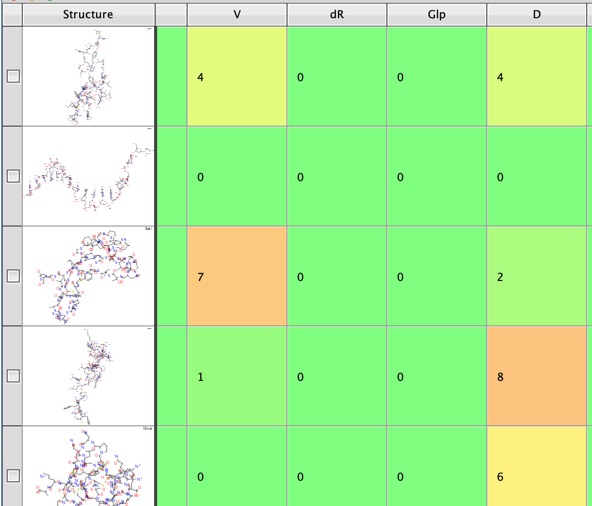
Once the substructure searching is complete the next part of the script generates a new workspace with a count of the number of peptides that contain each amino acid.
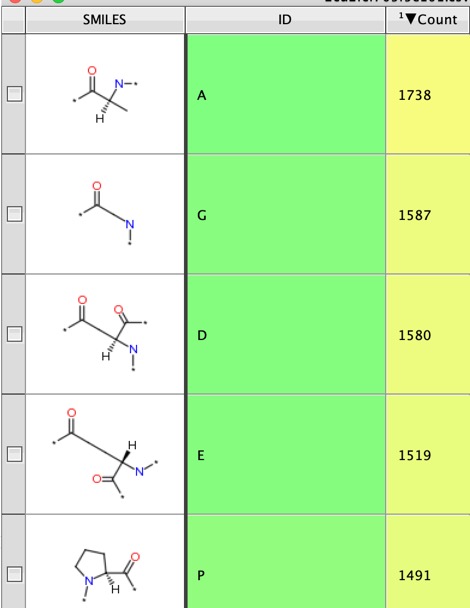
The script runs remarkably quickly, for a dataset of nearly 9000 peptides search for round 3000 amino-acids the whole process took around 4 mins.
The Vortex Script
# Provides a counter of amino-acids defined by SMARTS
# swain@mac.com
# do@dotmatics.com
#
import java
import re
import collections
from com.dotmatics.vortex.mol2img import Mol2Img
from Queue import Queue
from threading import Thread
processorcount = java.lang.Runtime.getRuntime().availableProcessors()
class smilesworker(Thread):
def __init__(self, q, eval_column):
self.q = q
self.eval_column = eval_column
Thread.__init__(self)
def run(self):
while 1:
row = self.q.get()
if row == None:
return
try:
vortex_tmp_value = vortex.getMolProperty(vtable.getStructureText(row), "SMILES")
except:
vortex_tmp_value = None
if (vortex_tmp_value == None):
self.eval_column.setValueFromString(row, None)
else:
self.eval_column.setValueFromString(row, str(vortex_tmp_value))
#SMARTS patterns here
patterns = []
#you will need to edit path to SMARTS,txt file
for line in open('/Users/username/vortex/scripts/My_Scripts/Macrocycles/SMARTS.txt'):
m = re.match("\('(.+?)', '(.+?)'\)", line)
patterns.append((m.group(1), m.group(2)))
class match_multiple(ProgressRunnable):
def __init__(self):
self.useMatchCount = 0
self.calcSMILES = False
self.nostructure = False
self.structureColumn = vtable.findColumnWithName("SMILES")
if self.structureColumn == None:
self.calcSMILES = True
#vortex.alert(str(self.calcSMILES))
#vortex.alert(str(vtable.findColumnWithName(vtable.MolfileColumn)))
if (self.calcSMILES == True ) & (vtable.findColumnWithName(vtable.MolfileColumn) == None):
vortex.alert("You need an SD file or a SMILES column")
self.nostructure = True
def doCalcSmiles(self):
self.structureColumn.setValueFromString(vtable.getRealRowCount() - 1, None)
q = Queue(processorcount * 20)
#The workers
t = []
#Create workers
for i in range(0, processorcount):
t.append(smilesworker(q, self.structureColumn))
#Start the workers
for i in range(0, processorcount):
t[i].start()
#Load the Q
for row in range(0, vtable.getRealRowCount()):
q.put(row)
#Something to sell the workers to stop
for i in range(0, processorcount):
q.put(None)
for i in range(processorcount):
t[i].join()
def updateProgress(self, perc, message):
self.setProgressValue(perc)
self.setProgressMessage(message)
def run(self):
if not self.nostructure:
self.updateProgress(0, 'Calculating SMILES')
if (self.calcSMILES):
self.structureColumn = vtable.findColumnWithName("SMILES", 1, vortex.STRING)
self.doCalcSmiles()
self.updateProgress(0, 'Indexing SMILES (for performance)')
Mol2Img.doSearch(self.structureColumn, '[U].Cl.F.Br.N.O.S', 'nomdl', 1)
results = []
for i in range(0, vtable.getRealRowCount()):
results.append([])
message = ''
ttotal = 0
for i in range(0, len(patterns)):
self.updateProgress(int(100 * (float(i) / float(len(patterns)))), patterns[i][0])
hits = Mol2Img.doSearch(self.structureColumn, patterns[i][1], 'nomdl', 1)
mycol = vtable.findColumnWithName(patterns[i][0], 1, vortex.INT)
for i in range(vtable.getRealRowCount()):
if hits.containsKey(i):
mycol.setInt(i, hits[i])
else:
mycol.setInt(i, 0)
vtable.fireTableStructureChanged()
if vws is None:
vortex.alert("You must have a workspace loaded...")
else:
matcher = match_multiple()
vortex.run(matcher, "Generating matches")
#Generate new workspace with summary of AA
newcols = ["ID", "SMILES", "Count"]
patterns2 = [list(a) for a in patterns]
# patterns2 format [['X2484', '*C(=O)[C@@]([H])(N(*)[H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])SC(=O)C([H])([H])[H]'],
results = []
for c in patterns2:
SelColumn = vtable.findColumnWithName(c[0],1)
colscore = 0
rows = vtable.getRealRowCount()
for r in range(0, int(rows)):
molScore = SelColumn.getValue(r)
if molScore > 0:
colscore = colscore + 1
c.append(colscore)
results.append(c)
arrayToWorkspace(results, newcols, 'TestAA summary')
The Jupyter Notebook, SMARTS.txt and Vortex script can be downloaded here
Last updated 29 August 2019