A little while back I described a docking workflow including a rescoring script for Vortex, so I thought it might be useful to include this on a separate page.
Recently, machine-learning scoring functions trained on protein-ligand complexes have shown significant promise an example being (RF-Score-VS) trained on 15 426 active and 893 897 inactive molecules docked to a set of 102 targets DOI.
Our results show RF-Score-VS can substantially improve virtual screening performance: RF-Score-VS top 1% provides 55.6% hit rate, whereas that of Vina only 16.2% (for smaller percent the difference is even more encouraging: RF-Score-VS top 0.1% achieves 88.6% hit rate for 27.5% using Vina). In addition, RF-Score-VS provides much better prediction of measured binding affinity than Vina (Pearson correlation of 0.56 and −0.18, respectively). Lastly, we test RF-Score-VS on an independent test set from the DEKOIS benchmark and observed comparable results.
Binaries for RF-Score-VS are available https://github.com/oddt/rfscorevs_binary and requires only minimal input
- -i input file format; if not present then based on extension [optional]
- –receptor a protein file; format based on extension [required]
- -O output file; if -o is not present file format is based on extension [optional]
- -o output file format; if -O is not present then molecules are printed to standard output [optional]
Thus the command line is
'/usr/local/bin/rf-score-vs_v1/rf-score-vs', '--receptor', pdbFile, sdfFile, '-o', 'csv', '--field', 'name', '--field', 'RFScoreVS_v2'
This can be accessed via a Vortex script, first we get the path to the imported SDF file, then use a dialog box to get the path to the PDB file used for the docking. Then we construct the command for rescoring and submit it. Finally we parse the output returned and populate the workspace.
Vortex script for rescoring docking results
#script to rescore binding results
# Performance of machine-learning scoring functions in structure-based virtual screening
# Maciej Wjcikowski, Pedro J. Ballester & Pawel Siedlecki
# doi:http://dx.doi.org/10.1038/srep46710
import sys
import os
import subprocess
# Get the path to the currently open sdf file
sdfFile = vortex.getFileForPropertyCalculation(vtable)
#uncomment for testing
#if file:
# vortex.alert(str(sdfFile))
# Get path to PDB for docking
# Open a dialog to choose a file
# getFile(title, extensions, 0 = Open, 1 = Save)
# vortex will keep track of the last folder you looked in etc
file = vortex.getFile("Choose the pdb the ligands were docked into", [".pdb"], 0)
#display file path, uncomment for testing
#if file:
# vortex.alert(str(file))
pdbFile=file.getAbsolutePath()
#column = vtable.findColumnWithName('RFScoreVS_v2', 1, 3)
p = subprocess.Popen(['/usr/local/bin/rf-score-vs_v1/rf-score-vs', '--receptor', pdbFile, sdfFile, '-o', 'csv', '--field', 'name', '--field', 'RFScoreVS_v2'], stdout=subprocess.PIPE, stderr=subprocess.PIPE)
output = p.communicate()[0]
# Parse the output, first line names for column
lines = output.split('\r\n')
colName = lines[0].split(',')
for c in colName:
column = vtable.findColumnWithName(c, 1)
vtable.fireTableStructureChanged()
rows = lines[1:len(lines)]
for r in range(0, vtable.getRealRowCount()):
vals = rows[r].split(',')
for j in range(0, len(vals)):
column = vtable.findColumnWithName(colName[j], 0)
column.setValueFromString(r, vals[j])
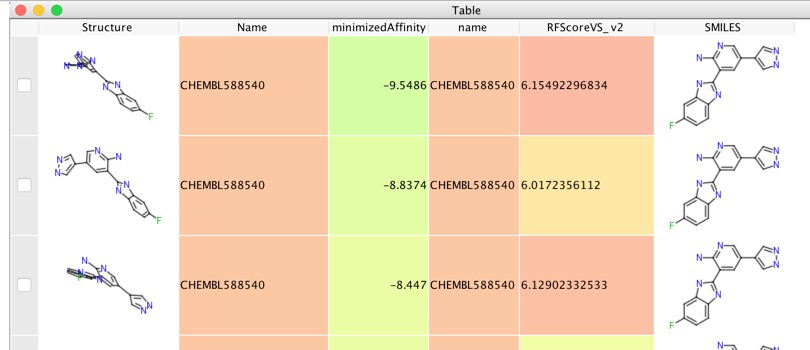
This script rescores all the docking poses and populates the table as shown below. Sometimes it can be a little difficult to discern the 3D structures displayed in Vortex so a useful trick is to click on the “Tools” menu and select “Calculate Properties”, then in the dialog box select the check box alongside “SMILES code of molecule”. An additional column will be added that contains the SMILES string rendered as a 2D molecule.