alvaDesc is a desktop tool for the calculation of a wide range of molecular descriptors and a number of molecular fingerprints from https://www.alvascience.com. alvaDesc can be used to determine over 5000 different descriptors (the full list is here). As might be expected it can be used to calculate the usual physicochemical properties such as molar refractivity, topological polar surface area {TPSA}, molecular volume estimations, two LogP models {Moriguchi and Ghose-Chippen octanol-water partition coefficient}. However there is no pKa calculation and no LogD calculation. There are a number of atom and ring count descriptors.
There is a significative list of drug-like and lead-like alerts including the well-known Lipinski rule of five, in addition to rules formulated by Oprea, Walters, Chen, Zheng, Rishton and Veber. There are also lead-like scoring from Congreave and Monge.
alvaDesc opens like any Mac application with a double click, there is also extensive help available from the help menu. You can then import a molecular file, I have a folder of sample files in different formats and it seems a wide range were recognised {SMILES, MDL, MOL, SDF, SD} but not {CDX, XYZ, CML, MDB}.
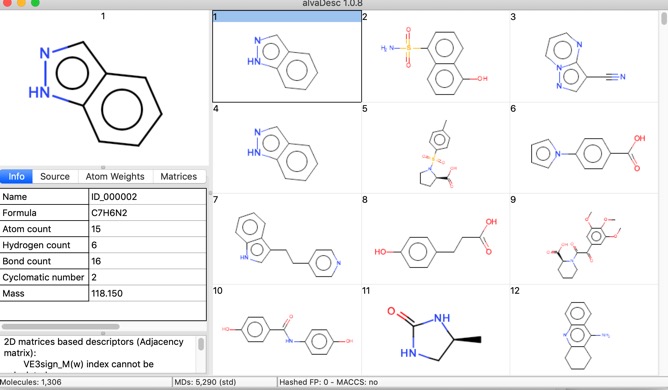
One thing I noticed is that when reading a file in SDF format whilst all the data fields are imported the Name is taken from the first title line of file record, this is not always populated, in these cases the “Name” is assigned as the filename followed by a number {e.g. publishedFragments_3}. It might be useful to be able to select an identifier field in the file. If you read in a file containing SMILES strings the name is the SMILES string.
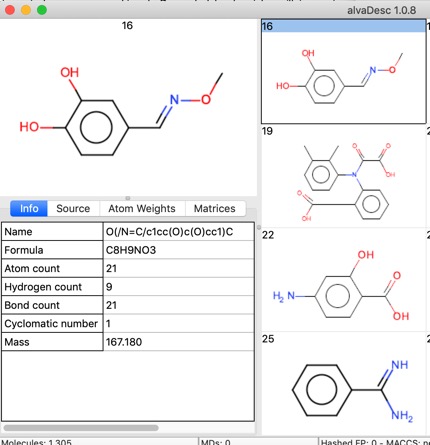
If the SDF file has a title then this is read into the “Name”.
A few properties are calculated on import such as Molecular Formula, Atom Count, Molecular Weight, and the “Source” Tab gives access to the record details including any data fields in the SDF file.
One of the common issues is that molecule structure files often contain disconnected fragments/counterions etc. alvaDesc can deal with not fully connected structures, such as salts, mixtures and ionic liquids. alvaDesc provides different strategies to calculated molecular descriptors on this kind of molecules, one of this option is the “Retain the biggest fragment approach” which means that the molecular descriptors are calculated only for the biggest fully connected structure included in the original molecule. In case of molecules with counter ions this option removes them from the structure.
Calculation of Descriptors
alvaDesc allows the calculation of a large number of possible descriptors, which can be selected from a menu.
Despite only using a single core on my machine alvaDesc calculates descriptors with impressive speed, calculating over 5000 descriptors for a 1300 molecule pdf took less than 50 seconds, I mentioned this to the developer and he sent an update the same day with multicore support!!
Once calculated, the descriptors can be exported to tab-separated text files by clicking File on the menu bar and choose Save descriptors.
The calculated descriptors can then be analysed in a number of ways by clicking Analysis on the menu bar
- Univariate statistics shows basic statistical data about the descriptors {mean, standard deviation, etc.}
- Principal Component Analysis can be used to perform the well-known PCA statistical procedure. At the end of the analysis the PCA Loadings and Scores plots will be shown
- Correlate analysis shows the relationship between n descriptors.
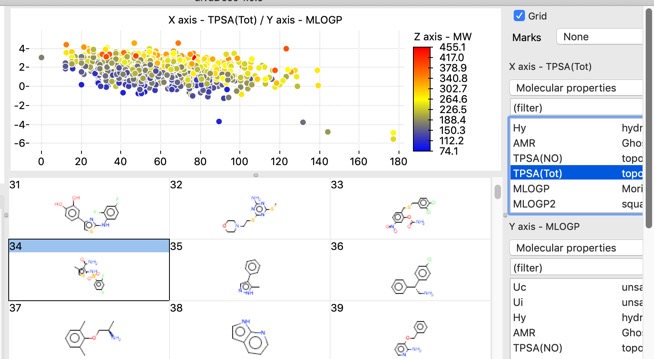
There are also a number of plotting features with in the app, histograms, bar charts and scatterplots.
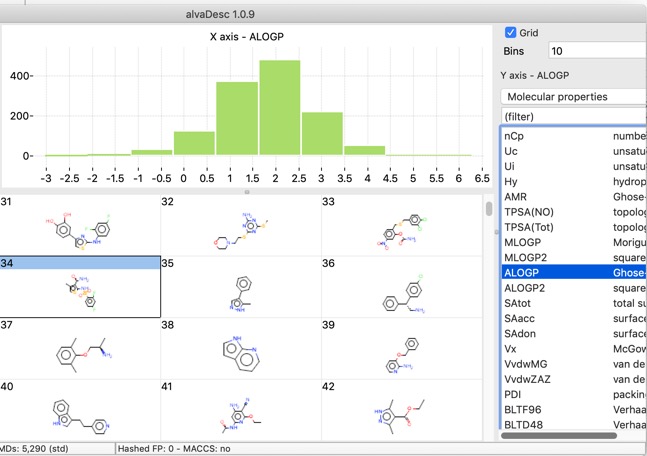
Scatter plots can also colour coded with a third property.
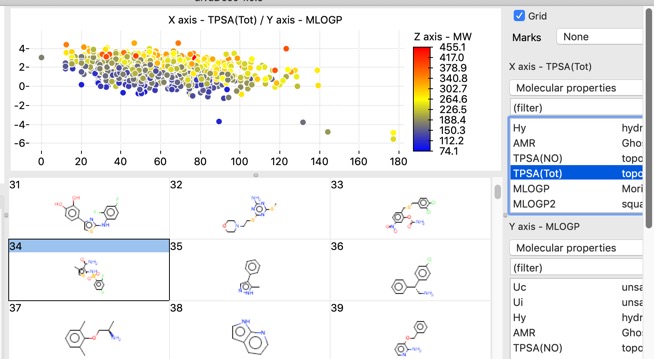
The chart points can either be selected individually, by clicking on each one, or as group by clicking whilst pressing SHIFT and dragging a rectangle around the items you want to select. This action will filter the molecules shown in the main window to those that are selected in the plot.
alvaDesc can also be used to calculate a number of fingerprints including MACCS, ECFP and pathfingerprints.
alvaDesc also has a scripting interface that allows automating import and descriptor/fingerprint calculation.
Accessing alvaDesc from the Commandline
alvaDesc can also be accessed from the command line, the executable is bundled within the the App {you can see it by right-click on the application icon and selecting show package contents}, you will need to either include the folder in you BASH profile or include the full path to the executable as shown below. You will also need to ensure you enclose and SMILES string within quotes.
/Applications/alvaDesc.app/Contents/MacOS/alvaDescCLI --iSMILES='CN(C)C1=C2C=CC=CC2=NC=C1' --descriptors=MW
alvaDescCLI.
Version 1.0 for Darwin (x86_64) - v.1.0.9 - built on: 2019/04/08
alvaDesc Commercial Single license. Expiration date: 2019/05/03
Licensee: Macs in Chemistry
Date: Sunday, 14 April 2019
Time: 10:50:45
172.25The advantage of the command line interface is that alvaDesc can be called by an external application to provide molecular descriptors or fingerprints, for example using a Jupyter Notebook as shown below, which uses RDKit to display the structures.
# Command format for alvaDesc
#/Applications/alvaDesc.app/Contents/MacOS/alvaDescCLI --input='/Users/username/Desktop/SampleFiles/PublishedFragments_test.sdf' --descriptors="MW,ALOGP"
from io import StringIO
import pandas as pd
from rdkit import Chem
from rdkit.Chem import AllChem
from numpy import linalg
from rdkit.Chem import PandasTools
import os
from rdkit import RDConfig
Path to SDF file containing structures
sdfFilePath = '/Users/username/Desktop/SampleFiles/PublishedFragments_test.sdf'
sdfFilePath
Use the command line alvaDesc
output = !/Applications/alvaDesc.app/Contents/MacOS/alvaDescCLI --input='{sdfFilePath}' --labels --descriptors="MW,ALOGP"
output = '\n'.join(output)
#output
Read output into pandas dataframe
df = pd.read_csv(StringIO(output), sep='\t')
df.head(n=10)
Remove rows containing no data
df=df.dropna()
df.head(n=4)
Read SDF file into pandas using RDKit
rddf = PandasTools.LoadSDF(sdfFilePath,molColName='Molecule')
rddf.head(n=3)
| ID | Molecule | |
|---|---|---|
| 0 | ID_000002 |  |
| 1 | ID_000003 |  |
| 2 | ID_000004 |  |
Merge dataframes using molecule name as key
resultCA_PBF = pd.merge(df, rddf, left_on='NAME', right_on='ID')
resultCA_PBF.head(n=3)
| No. | NAME | MW | ALOGP | ID | Molecule | |
|---|---|---|---|---|---|---|
| 0 | 1 | ID_000002 | 118.15 | 1.4738 | ID_000002 | |
| 1 | 2 | ID_000003 | 223.27 | 1.2992 | ID_000003 | |
| 2 | 3 | ID_000004 | 144.15 | 0.3616 | ID_000004 | |
alvaDesc is also available as set of KNIME nodes https://www.alvascience.com/knime-alvadesc/ but note in order to work, the plugin requires a licensed version of alvaDesc installed on the same computer. Last Updated 14 April 2019