I’m regularly handling very large files containing millions for chemical structures and whilst BBEdit is my usual tool for editing text files in practice it becomes rather cumbersome for really large files (> 2 GB). In these cases I’ve compiled a useful list of UNIX commands that make life easier. Whilst I use them when dealing with large chemical structure files they are equally useful when dealing with any large text or data files.
If you start up the Terminal application which is in the Utilities folder in the Applications folder. You should see a window something like this:-
The shell prompt (or command line) is where one types commands, the appearance may differ slightly depending on customisation.
File and Path Names
One thing to remember is that UNIX is less accommodating than Mac OS X when it comes to file name and the paths to files. When you see the full path to a file it will be shown as a list of directory names starting from the top of the tree right the way down to the level where your file sits. It will look something like this.
/Users/Username/Projects/bioconda-recipes/recipes/rdkit/2016.03.3/meta.yaml
Directory names can contain spaces but you’ll often find that people working in Unix prefer to keep both file and directory names without spaces since this simplifies the process of working with these files on a command line. Directory or file names containing a space must be specified on the command line either by putting quotes around them, or by putting a \ (backslash) before the space.
'/Users/Username/Projects/bioconda recipes/recipes/rdkit/2016.03.3/meta.yaml '
or
/Users/Username/Projects/bioconda\ recipes/recipes/rdkit/2016.03.3/meta.yaml
If typing out the full path seems rather daunting remember if you drag a file in the Finder and drop it on a Terminal window the path to the file will be automatically generated.
Examining Large files
The other thing to remember is that there is an extensive help available, so if you can’t remember the command line arguments “man” can provide the answers. Simply type man followed by the command.
MacPro:~ Chris$ man wc
WC(1) BSD General Commands Manual WC(1)
NAME
wc -- word, line, character, and byte count
SYNOPSIS
wc [-clmw] [file ...]
DESCRIPTION
The wc utility displays the number of lines, words, and bytes contained
in each input file, or standard input (if no file is specified) to the
standard output. A line is defined as a string of characters delimited
by a <newline> character. Characters beyond the final <newline> charac-
ter will not be included in the line count.
A word is defined as a string of characters delimited by white space
characters. White space characters are the set of characters for which
the iswspace(3) function returns true. If more than one input file is
specified, a line of cumulative counts for all the files is displayed on
a separate line after the output for the last file.
The following options are available:
-c The number of bytes in each input file is written to the standard
output. This will cancel out any prior usage of the -m option.
-l The number of lines in each input file is written to the standard
output.
-m The number of characters in each input file is written to the
standard output. If the current locale does not support multi-
byte characters, this is equivalent to the -c option. This will
cancel out any prior usage of the -c option.
-w The number of words in each input file is written to the standard
output.
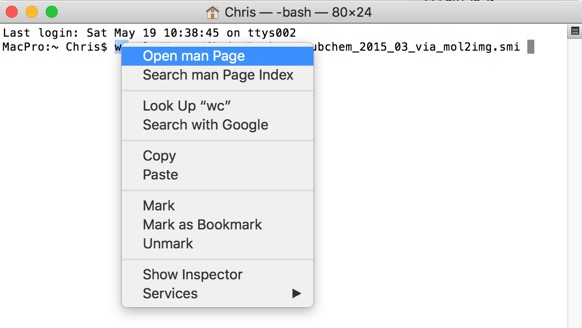
In addition to quickly view the man page for any command, just right-click on the command in the Terminal and choose Open man Page from the context menu. A new window will pop up, displaying the manual for that command.
So the command wc (word count) can be used to count words, lines, characters, or bytes depending on the option used. For example for a file containing SMILES strings the following command tells us how many lines (and hence the number of molecules). In this case the 4GB file contains 66,783,025 molecules.
MacPro:~ Chris$ wc -l /Users/Chris/Desktop/pubchem_2015_03_via_mol2img.smi
66783025 /Users/Chris/Desktop/pubchem_2015_03_via_mol2img.smi
If instead of counting lines we count words using the -w option, we get a different answer. This is because SMILES string can contain salts, counterions etc.
MacPro:~ Chris$ wc -w /Users/Chris/Desktop/pubchem_2015_03_via_mol2img.smi
133566050 /Users/Chris/Desktop/pubchem_2015_03_via_mol2img.smi
All the structures from ChEMBL are available for download as a 3.5 GB sdf file. Attempting to open such a file in a desktop text editor would not be recommended, however using a few UNIX commends we can interrogate the file to get useful information. For example the following tells us how many lines there are in the file.
Chris$ wc -l /Users/Chris/Desktop/chembl_22.sdf
119893133 /Users/Chris/Desktop/chembl_22.sdf
However in this case since each molecule record in a sdf file has multiple lines we don’t know how many structures there are. We can however look at a small portion of the file using the head command
MacPro:~ Chris$ man head
HEAD(1) BSD General Commands Manual HEAD(1)
NAME
head -- display first lines of a file
SYNOPSIS
head [-n count | -c bytes] [file ...]
DESCRIPTION
This filter displays the first count lines or bytes of each of the speci-
fied files, or of the standard input if no files are specified. If count
is omitted it defaults to 10.
So if we type
MacPro:~ Chris$ head -50 /Users/Chris/Desktop/chembl_22.sdf
CHEMBL153534
SciTegic12231509382D
16 17 0 0 0 0 999 V2000
7.6140 -22.2702 0.0000 C 0 0
5.7047 -23.1991 0.0000 C 0 0
6.1806 -22.5282 0.0000 N 0 0
6.9604 -22.7690 0.0000 C 0 0
4.8790 -23.2163 0.0000 N 0 0
8.2791 -21.1119 0.0000 N 0 0
7.5280 -21.4445 0.0000 C 0 0
8.4225 -22.4364 0.0000 C 0 0
8.8353 -21.7198 0.0000 C 0 0
6.2035 -23.8527 0.0000 S 0 0
4.0534 -23.2163 0.0000 C 0 0
6.9776 -23.5889 0.0000 C 0 0
3.6406 -22.4938 0.0000 N 0 0
3.6406 -23.9215 0.0000 N 0 0
8.4397 -20.3035 0.0000 C 0 0
9.6552 -21.6280 0.0000 C 0 0
2 3 2 0
3 4 1 0
4 1 1 0
5 2 1 0
6 7 1 0
7 1 2 0
8 1 1 0
9 8 2 0
10 12 1 0
11 5 2 0
12 4 2 0
13 11 1 0
14 11 1 0
15 6 1 0
16 9 1 0
6 9 1 0
10 2 1 0
M END
> <chembl_id>
CHEMBL153534
$$$$
CHEMBL440060
11280716012D 1 1.00000 0.00000 0
206208 0 1 0 999 V2000
2.0792 -0.9500 0.0000 N 0 0 0 0 0 0 0 0 0
1.4671 -0.5958 0.0000 C 0 0 2 0 0 0 0 0 0
0.8510 -0.9500 0.0000 C 0 0 0 0 0 0 0 0 0
0.8510 -1.6583 0.0000 O 0 0 0 0 0 0 0 0 0
We can now see that each molecule record is separated by $$$$ and we can use this to identify how many molecules are in the file using grep and counting the number of occurrences.
MacPro:~ Chris$ man grep
SYNOPSIS
grep [-abcdDEFGHhIiJLlmnOopqRSsUVvwxZ] [-A num] [-B num] [-C[num]]
[-e pattern] [-f file] [--binary-files=value] [--color[=when]]
[--colour[=when]] [--context[=num]] [--label] [--line-buffered]
[--null] [pattern] [file ...]
DESCRIPTION
The grep utility searches any given input files, selecting lines that
match one or more patterns. By default, a pattern matches an input line
if the regular expression (RE) in the pattern matches the input line
without its trailing newline. An empty expression matches every line.
Each input line that matches at least one of the patterns is written to
the standard output.
So if we type this in the terminal window.
MacPro:~ Chris$ grep -c '$$$$' /Users/Chris/Desktop/chembl_22.sdf
1678393
In a similar manner we can use tail to look at the end of the file.
MacPro:~ Chris$ tail -n10 /Users/Chris/Desktop/chembl_22.sdf
22 24 1 0
8 20 1 0
8 17 1 0
54 55 2 0
8 60 1 6
M END
> <chembl_id>
CHEMBL3706412
$$$$
Sometimes you just want to test a new tool on a subset of a large file, in this case we can use head and pipe the results into a new file.
MacPro:~ Chris$ head -10000 /Users/Chris/Desktop/pubchem_2015_03_via_mol2img.smi > /Users/Chris/Desktop/UnixLargeFiles/test.txt
Dealing with Errors in very large files
Most command line applications expect the file impute to have unix style line endings, whilst most modern applications use unix style ending if you are using an old file it may have the old style Mac line endings, or dos line endings. There is a very useful tool to convert to unix line endings, dos2unix can be installed using home-brew
brew install dos2unix
And then used with the following command.
dos2unix /Users/username/Desktop/testsmi.smi
Sometimes a file contains blank lines that need removing, this is easy to do using grep
grep -v '^$' /Users/username/Desktop/testsmi.smi > /Users/chrisswain/Desktop/testsmi2.smi
Sometimes files contain non-printable ASCII characters and it can be hard to track these down. Whilst you can use BBEdit to zap these characters but for larger files this command might be a better option.
You can use this Perl command to strip these characters by piping your file through it:
perl -pi.bak -e 's/[\000-\007\013-\037\177-\377]//g;'
If that does not work the best option is often to simply split a file into smaller files
MacPro:~ Chris$ man split
-l line_count
Create smaller files n lines in length.
-p pattern
The file is split whenever an input line matches pattern, which
is interpreted as an extended regular expression. The matching
line will be the first line of the next output file. This option
is incompatible with the -b and -l options.
If additional arguments are specified, the first is used as the name of
the input file which is to be split. If a second additional argument is
specified, it is used as a prefix for the names of the files into which
the file is split. In this case, each file into which the file is split
is named by the prefix followed by a lexically ordered suffix using
suffix_length characters in the range We can split the file test.txt created earlier
MacPro:~ Chris$ split -l 100 /Users/Chris/Desktop/UnixLargeFiles/test.txt /Users/Chris/Desktop/UnixLargeFiles/splitfile
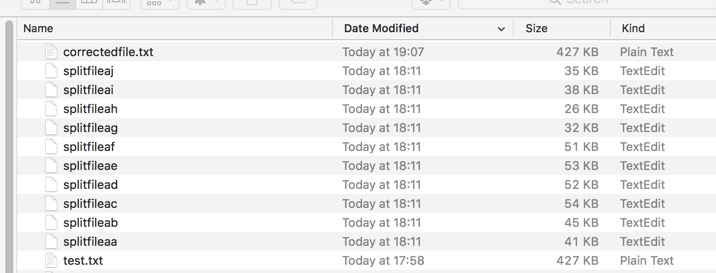
You can then use wc to check the concatenated file has the correct number of lines.
Editing Files
cat is also the swiss army knife for file manipulation, combined with tr
MacPro:~ Chris$ man tr
TR(1) BSD General Commands Manual TR(1)
NAME
tr -- translate characters
SYNOPSIS
tr [-Ccsu] string1 string2
tr [-Ccu] -d string1
tr [-Ccu] -s string1
tr [-Ccu] -ds string1 string2
DESCRIPTION
The tr utility copies the standard input to the standard output with substitution or deletion of selected characters.
To convert a tab delimited file into commas
cat tab_delimited.txt | tr "\\t" "," comma_delimited.csv
For some toolkits the SMILES file containing structures needs to have UNIX line endings, if your input file does not have UNIX line endings you can either use a text editor like BBEdit to change it to UNIX format of run this command in the Terminal
cat input_file.txt | tr '\r' '\n' | tr -s '\n' > unix_file.txt
Monitoring output files
The command tail has two special command line option -f and -F (follow) that allows a file to be monitored. Instead of just displaying the last few lines and exiting, tail displays the lines and then monitors the file. As new lines are added to the file by another process, tail updates the display. This very useful if you are monitoring a log file to check for errors when processing a very large file.
tail -f /Users/Chris/Desktop/UnixLargeFiles/output.log
Checking for duplicate structures
One of the issues with combining multiple data sets is there is always the risk of duplicate structures. In order to check for this you need a unique identifier for each molecular structure, I tend to use InChIKey.
The IUPAC International Chemical Identifier InChI is a textual identifier for chemical substances, designed to provide a standard way to encode molecular information and to facilitate the search for such information in databases and on the web. The condensed, 27 character InChIKey is a hashed version of the full InChI (using the SHA-256 algorithm).
First use OpenBabel to generate the InChiKey file
MacPro:~ Chris$ /usr/local/bin/babel -ismi '/Users/Chris/Desktop/UnixLargeFiles/withdupsfile.smi' -oinchikey '/Users/Chris/Desktop/UnixLargeFiles/output.inchikey'
We can check the file using head
MacPro:~ Chris$ head -10 /Users/Chris/Desktop/UnixLargeFiles/output.inchikey
RDHQFKQIGNGIED-UHFFFAOYSA-N
RDHQFKQIGNGIED-UHFFFAOYSA-O
INCSWYKICIYAHB-UHFFFAOYSA-N
HXKKHQJGJAFBHI-UHFFFAOYSA-N
HIQNVODXENYOFK-UHFFFAOYSA-N
VYZAHLCBVHPDDF-UHFFFAOYSA-N
MUIPLRMGAXZWSQ-UHFFFAOYSA-N
PDGXJDXVGMHUIR-UHFFFAOYSA-N
INAPMGSXUVUWAF-UHFFFAOYSA-N
AUFGTPPARQZWDO-UHFFFAOYSA-N
We now need to sort the file
MacPro:~ Chris$ sort /Users/Chris/Desktop/UnixLargeFiles/output.inchikey -o /Users/Chris/Desktop/UnixLargeFiles/sortedoutput.inchikey
Now we can use uniq to identify duplicate structures, it is important to note uniq filters out adjacent, matching lines which is why we need to sort first.
MacPro:~ Chris$ uniq -c /Users/Chris/Desktop/UnixLargeFiles/sortedoutput.inchikey
1 AAAFZMYJJHWUPN-UHFFFAOYSA-N
1 AAALVYBICLMAMA-UHFFFAOYSA-N
1 AANFHDFOMFRLLR-UHFFFAOYSA-N
3 AAOVKJBEBIDNHE-UHFFFAOYSA-N
1 AAQOQKQBGPPFNS-UHFFFAOYSA-N
1 AAWZDTNXLSGCEK-UHFFFAOYSA-N
1 AAZMHPMNAVEBRE-UHFFFAOYSA-M
1 AAZMHPMNAVEBRE-UHFFFAOYSA-N
1 ABBADGFSRBWENF-UHFFFAOYSA-N
2 ABCOOORLYAOBOZ-UHFFFAOYSA-N
2 ABETXLPRGREJND-UHFFFAOYSA-M
Alternative options for uniq are
-d Only output lines that are repeated in the input.
-u Only output lines that are not repeated in the input.
Extracting data from delimited files
Sometimes you have very large files containing multiple columns of data, but you really want is a single column. The file is too big to open in a spreadsheet application but you can extract just the column you want using a simple command. First use head to identify the column of data you want.
MacPro:~ Chris$ head -3 /Users/Chris/Desktop/UnixLargeFiles/alldata.csv
StructureID,chembl_id,MW,XLogP,HBA,HBD,HAC,RotBondCount,SMILES
0,CHEMBL153534,235.31,1.97,5,2,16,2,Cc1cc(cn1C)-c1csc(n1)N=C(N)N
978,CHEMBL440060,2883.34,-12.48,78,40,202,95,CC[C@H](C)[C@H](NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](NC(=O)[C@@H](N)CCSC)[C@@H](C)O)C(=O)NCC(=O)N[C@@H](C)C(=O)N[C@@H](C)C(=O)N[C@@H](Cc1c[nH]cn1)C(=O)N[C@@H](CC(=O)N)C(=O)NCC(=O)N[C@@H] (C)C(=O)N[C@@H](C)C(=O)N[C@@H](CCC(=O)N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCN=C(N)N)C(=O)N[C@@H](CCC(=O)N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCN=C(N)N)C(=O)NCC(=O)N[C@@H](CCC(=O)N)C(=O)N[C@@H] (CC(C)C)C(=O)NCC(=O)N1CCC[C@H]1C(=O)N1CCC[C@H]1C(=O)NCC(=O)N[C@@H](CO)C(=O)N[C@@H](CCCN=C(N)N)C(=O)N
The column we want is SMILES, which is column 9. We can use cat to extract just this column (replace f9 with the column you want).
MacPro:~ Chris$ cat /Users/Chris/Desktop/UnixLargeFiles/alldata.csv |cut -d ',' -f9
SMILES
Cc1cc(cn1C)-c1csc(n1)N=C(N)N
CC[C@H](C)[C@H](NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](NC(=O)[C@@H](N)CCSC)[C@@H](C)O)C(=O)NCC(=O)N[C@@H](C)C(=O)N[C@@H](C)C(=O)N[C@@H](Cc1c[nH]cn1)C(=O)N[C@@H](CC(=O)N)C(=O)NCC(=O)N[C@@H](C)C(=O)N[C@@H](C)C(=O)N[C@@H] (CCC(=O)N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCN=C(N)N)C(=O)N[C@@H](CCC(=O)N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCN=C(N)N)C(=O)NCC(=O)N[C@@H](CCC(=O)N)C(=O)N[C@@H] (CC(C)C)C(=O)NCC(=O)N1CCC[C@H]1C(=O)N1CCC[C@H]1C(=O)NCC(=O)N[C@@H](CO)C(=O)N[C@@H](CCCN=C(N)N)C(=O)N
If you want to save the output to a file
MacPro:~ Chris$ cat /Users/Chris/Desktop/UnixLargeFiles/alldata.csv |cut -d ',' -f9 > /Users/Chris/Desktop/UnixLargeFiles/justSMILES.smi
Dealing with a large number of files
A suggestion from a reader. Sometimes rather than one large file download sites provide the data as a large number of individual files. We can keep track of the number of files using this simple command. In my testing this works fine, but will not count hidden files, and will miscount files that contain space/line breaks etc. in file names.
MacPro:~ Chris$ ls | wc -1
177248
xsv is a command line program for indexing, slicing, analyzing, splitting and joining CSV files. Commands should be simple, fast and composable:
Available commands
cat – Concatenate CSV files by row or by column.
count – Count the rows in a CSV file. (Instantaneous with an index.)
fixlengths – Force a CSV file to have same-length records by either padding or truncating them.
flatten – A flattened view of CSV records. Useful for viewing one record at a time. e.g., xsv slice -i 5 data.csv | xsv flatten.
fmt – Reformat CSV data with different delimiters, record terminators or quoting rules. (Supports ASCII delimited data.)
frequency – Build frequency tables of each column in CSV data. (Uses parallelism to go faster if an index is present.)
headers – Show the headers of CSV data. Or show the intersection of all headers between many CSV files.
index – Create an index for a CSV file. This is very quick and provides constant time indexing into the CSV file.
input – Read CSV data with exotic quoting/escaping rules.
join – Inner, outer and cross joins. Uses a simple hash index to make it fast.
partition – Partition CSV data based on a column value.
sample – Randomly draw rows from CSV data using reservoir sampling (i.e., use memory proportional to the size of the sample).
reverse – Reverse order of rows in CSV data.
search – Run a regex over CSV data. Applies the regex to each field individually and shows only matching rows.
select – Select or re-order columns from CSV data.
slice – Slice rows from any part of a CSV file. When an index is present, this only has to parse the rows in the slice (instead of all rows leading up to the start of the slice).
sort – Sort CSV data.
split – Split one CSV file into many CSV files of N chunks.
stats – Show basic types and statistics of each column in the CSV file. (i.e., mean, standard deviation, median, range, etc.)
table – Show aligned output of any CSV data using elastic tabstops.
Dividing large files
Sometimes it is useful to divide very large files into more manageable chunks. For SMILES files where we have one record per line we can simply divide based on lines using split.
SPLIT(1) BSD General Commands Manual SPLIT(1)
NAME split — split a file into pieces
SYNOPSIS split [-a suffixlength] [-b bytecount[k|m]] [-l line_count] [-p pattern] [file [name]]
split -l 10000 /Users/username/Desktop/SampleFiles/testfile.smi /Users/username/Desktop/SampleFiles/splitfile
Dividing sdf files is more problematic since we need each division to be at the end of a record defined by “$$$$”. I’ve spent a fair amount of time searching for a high-performance tool that will work for very, very large files. Many people suggest using awk
AWK (awk) is a domain-specific language designed for text processing and typically used as a data extraction and reporting tool. Like sed and grep, it’s a filter, and is a standard feature of most Unix-like operating systems.
I’ve never used awk but with much cut and pasting from the invaluable Stack Overflow this script seems to work.
awk -v RS='\\$\\$\\$\\$\n' -v nb=1000 -v c=1 '
{
file=sprintf("%s%s%06d.sdf",FILENAME,".chunk",c)
printf "%s%s",$0,RT > file
}
NR%nb==0 {c++}
' /Users/username/Desktop/SampleFiles/HitFinder_V11.sdf
The result is shown in the image below. There are a couple of caveats, this script only works with the version of awk shipped with Big Sur (you should be able to install gawk using Home Brew and use that on older systems), and it requires the file has unix line endings. The resulting file names is not ideal and if there are any awk experts out there who could tidy it up I’d be delighted to hear from you.
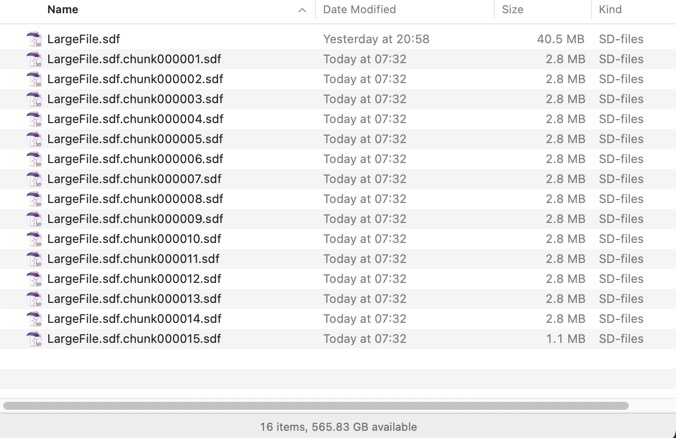
If anyone has any additional suggestions please feel free to submit them.
Last updated 18 Feb 2023